Section 8
Field Screening Methods
Interim Final – July, 2017
Section 8.0 Introduction
Section 8.1 Selecting a Field Screening Method
Section 8.2 Data Quality Control and Documentation
Section 8.3 Field Screening Methods and Documentation
Section 8.4 Field Screening Methods for Selected Contaminants and Media
8.4.1 Metals
8.4.2 Petroleum
8.4.3 PCBs
8.4.4 Dioxins
8.4.5 Volatiles
8.4.6 Pesticides and Herbicides
8.4.7 Explosives
Section 8.5 Field Screening With Cone Penetrometer and Sensors/ Probes
8.5.1 Description of Cone Penetrometer Technologies
8.5.2 Cone Penetrometer Data Verification
8.5.3 CPT Advantages and Limitations
8.5.4 Other CPT Instruments
Section 8.6 Field Screening Equipment to Support Health and Safety Programs
8.6.1 Combustible Gas Indicator (CGI)
8.6.2 Oxygen Meter
8.6.3 Flame Ionization Detector (FID)
8.6.4 Photoionization Detector (PID)
Figures
8-1 In situ and Ex situ Analysis of Soil Using Portable XRF
8-2 Use of Portable XRF for Ex situ Soil Analysis at an Industrial Site
8-3 Map of Total Arsenic Concentrations in Discrete Surface Soil Samples at a Former Pesticide-mixing Site
8-4 RemScan Handheld Infrared Spectrophotometry for Testing of TPH in Soil
8-5 RemediAid Colorimetry Test Kit for TPH in Soil
8-6 (A)PCB Test Kit Components and (B)RaPID Assay Immunoassay PCB Test Kits
8-7 Schematic of Sample Processing Steps for Use of CALUX Bioassay Kit for Dioxins and PCBs
8-8 PID for VOC screening
8-9 Container Setup for Headspace Screening
8-10 Pesticide Immunoassay Test Kit Components
8-11 Explosives Immunoassay Kit and Explosives Sample Analysis Using an Immunoassay Kit
8-12 Membrane Interface Probe with Conductivity Probe Tip
8-13 Combustible Gas Indicator
8-14 Oxygen Meter
8-15 Flame Ionization Detector
8-16 Photoionization Detector (MiniRAE)
Tables
8-1 Data Quality Levels for Field Screening Methods
8-2 Sources for Field Screening Methods
Return to the Top of the Page
8.0 FIELD SCREENING METHODS
The Hawai‘i Department of Health (HDOH) Hazard Evaluation and Emergency Response Office (HEER Office) supports the use of field screening methods to help streamline the site investigation or remediation process. This Section provides a general overview of selected field screening methods that have been approved by USEPA or other state environmental protection programs and is not intended to be comprehensive. A number of companies market field screening kits or instruments based on the methods described below or similar methods developed by a specific company. Alternative methods should be discussed with the HEER office on a site-by-site basis. Periodic updates on field screening methods will be included in this TGM Section. Detailed information is not provided on analytical systems used in conjunction with field screening on direct-push field platforms, including laser-produced fluorescence, membrane interface probes and fiber-optic chemical sensors. Refer to the USEPA link for Field Analytic Technologies noted in Table 8-2 for more detailed information on these methods.
The use of field screening methods is consistent with the “Triad” approach to site investigations promoted by the USEPA (USEPA, 2010e, Triad Resource Center, 2011). This includes the use of real-time field measurements to guide an investigation or remedial action, and optimize available resources as well as reduce the need for multiple remobilizations. Field screening should follow the same systematic planning steps outlined in Section 3 of the TGM. Site investigation objectives and decision statements should be developed to guide the investigation. This should include a thorough review of the site history to identify areas for targeted collection of field data, in addition to visual clues such as soil staining, obvious disposal areas, etc. Fixed laboratory data are generally collected for final decision making purposes. As described in this section, benefits of field screening include:
- Identification of contaminants of potential concern;
- Identification of areas of relative higher and lower contamination and the general magnitude of contamination in order to assist in initial site evaluation and Decision Unit (DU) designation for more detailed characterization;
- Rapid identification for removal of heavily contaminated soil prior to confirmation sampling using DU and Multi-Increment Sample (MIS) investigation approaches;
- Assessment of contaminant variability at the scale of individual sample points in order to estimate and optimize the mass and number of increments for Multi Increment (MI) sample collection;
- Selection of samples for initial laboratory analysis;
- Pre-screening of samples to optimize selection of laboratory analysis method (e.g. identification of low- or high-concentrations samples for the laboratory); and
- Carry out health and safety monitoring to determine possible worker hazards or exposures (generally not overseen by HDOH).
Field screening methods generate qualitative data (e.g., presence or absence),semi-quantitative data (e.g., above or below a pre-specified concentration), or quantitative data (specific concentration of targeted chemicals), depending on the method employed. It is important to understand both the advantages and limitations of the methods discussed. Field screening can be carried out in situ, without disturbing the targeted soil, or ex situ, on samples collected from targeted areas. Ex situ screening has traditionally been carried out on discrete samples but may also be used to screen MI samples (e.g., field XRF). The time and cost required to meet necessary data quality requirements for final decision making purposes utilizing field screening methods should be considered as part of the site investigation planning and site data quality objectives.
Note that a single test on a small mass of soil (e.g., one to ten grams) is unlikely to generate a representative mean concentration for either the bulk sample collected or the soil immediately surrounding a sampling point (refer to TGM Sections 3 and 4). Screening data for individual sample points should be used in a semi-quantitative manner, with the understanding that concentrations of the targeted COPCs in immediately surrounding soils could vary by up to an order of magnitude or more. The objective of screening is not to identify the “maximum” concentration of the COPC in the bulk sample or DU, since this is entirely dependent on the volume/mass of soil tested. Testing a small number of the likely millions of potential small masses of soil at a site will not identify the maximum concentration of the COPC present, and doing so is not relevant to investigation objectives.
The quality of field screening data will depend on representativeness of the sample tested and the method used. Higher quality field screening that generates concentrations of targeted contaminants is sometimes referred to as is “Field analysis” (see Subsection 8.2). Field screening should always be carried out in terms of “DUs” and well-thought-out DU questions and objectives, even if MI samples will not be collected at this point in the investigation (refer to Section 3). Testing of small masses of soil from unprocessed discrete samples, for example, are highly prone to error due to random small-scale distributional heterogeneity and variability of contaminant concentrations within any given mass of soil, including individual samples (see Subsection 4.1.1). This and other factors discussed below (e.g., soil moisture, particle size differences) can lead to “false negatives” or erroneous “cold spots” that lead to an underestimation of the extent and magnitude of contamination. Processing samples prior to screening when practical (e.g., drying and sieving prior to field XRF analysis) can help address these types of errors. However, the data are still subject to small-scale variability and erroneous decisions regarding apparently isolated “hot spots” and “cold spots.” At the other end of the spectrum, field XRF analysis of processed MI samples with accompanying field QA/QC by trained and experienced persons can generate high-quality data that rivals or exceeds laboratory data (refer to case study in Subsection 8.4.1). High quality data can also be obtained using field Gas Chromatography (GC). This could include the preservation of MI samples from excavation sidewalls or targeted intervals within borehole cores in methanol and analysis in the field to guide additional remediation or testing.
While large-scale patterns might be discernable from grid point sample data, boundaries between areas of “clean” and “contaminated” soil can be difficult to accurately establish based on traditional soil data (see Section 4). The zone between clean and heavily contaminated soil in particular is typically marked by scattered seemingly isolated “cold spots” and “hot spots” at the scale of a discrete sample (refer to XRF case study in Subsection 8.4.1. It is highly probable that this reflects random, small-scale variability of contaminant concentrations in the soil. If a new and independent set of discrete samples was collected across the same area, a similar pattern of cold spots and hot spots may appear, but in different locations. It is therefore important not to over interpret individual “cold spots” and “hot spots” identified with discrete sample data. Removal of soil in the immediate vicinity of apparent “hot spots” identified by a single or even a small number of discrete sample points is unlikely to reduce the average concentration across the area as a whole (see Section 4 for more detail on discrete sample variability). Properly designated DU-MIS soil samples are necessary to confirm initial estimates of the extent and magnitude of contamination based on grid point screening data.
This same type of random, distributional heterogeneity can introduce error in attempts to collect “splits” of samples for comparison of field screening data and fixed-laboratory data. Laboratory data may not necessarily be more accurate or representative of the original bulk sample if the sample handling and processing are not conducted appropriately. A significant and random variability between field and lab data could simply reflect inadequate processing of a sample before the splits were prepared. Refer to TGM Section 4 for additional information on subsampling of bulk samples for separate analyses.
When understood in this context, even qualitative and semi-quantitative screening data based on testing of samples from grid points designated across a targeted area could still be useful for the initial estimation of “clean” and “contaminated” areas, and improve the efficiency of more detailed followup investigations. Use of a field XRF by experienced personnel as discussed below is one example. Multi Increment samples tested at a fixed-laboratory are recommended to confirm initial decisions (see Section 4). Final confirmation could in theory be accomplished with field screening data provided that the following criteria are met: 1) An adequate number of increments (for MI samples) or alternative types of samples (e.g., discrete) of proper shape and mass are collected (see TGM Section 4 discussion of increment mass and shape), 2) The samples are assigned to well-thought-out DUs that include consideration of risk to human health and the environment as well as potential removal or remedial actions (refer to TGM Section 3), 3) Field QA/QC methods are comparable to QA/QC methods that are used at a fixed laboratory, 4) An adequate correlation of field screening data versus fixed-laboratory data is accomplished, and 5) Data for a subset of DUs are verified by replicate/independent sets of samples (refer to TGM Subsection 4.2.7).
The USEPA maintains a detailed overview of Field Analytic Techniques (USEPA, 2007). Refer to EPA’s web page for additional information. New methods are constantly being developed and can be discussed with HDOH for site application. Field screening methods for site investigation and cleanup are typically followed by or used in conjunction with laboratory analysis testing for decision-making. Used in concert with laboratory analysis measurements, field screening data can serve to expedite site characterization or site remediation activities and reduce overall costs.
8.1 SELECTING A FIELD SCREENING METHOD
There are a number of important considerations that must be taken into account when proposing a field screening method. This list includes items that should be evaluated (at a minimum) for each project:
- Target Analytes: Field screening methods generally provide results for either a specific compound or a specific class of compounds. For example, if the site contaminant is petroleum, but the specific concern is polynuclear aromatic hydrocarbons (PAH), select a method that targets PAHs, not just petroleum hydrocarbons. Additionally, the available field screening methods may not have adequate selectivity to test for specific compounds within a class of chemicals (e.g., individual PAHs or PCB isomers).
- Action Levels: Field screening methods may have higher detection limits than laboratory methods and some methods may not be sensitive enough to meet applicable action levels. Field methods should have adequate sensitivity to meet the goals and objectives established in the site SAP/QAPP.
- Matrix Effects: Some field screening methods may have limited usefulness with certain sample matrices. An example of matrix effects is soil with a high clay or organic carbon content, which can cause an immunoassay test to be biased low. Other examples of matrix effects include moisture or the presence of large pebbles in soil being screened with a field XRF. A preliminary study using soil from the targeted site is recommended prior to the beginning of field work in order to evaluate potential limitations of the proposed field screening methods. Calibration of equipment with site-specific or soil type-specific samples/standards and other methods recommended by the field screening test kit or instrument manufacturer can compensate for some of these effects.
- Data Use: Field screening methods generate a wide range of data quality, from purely qualitative data (e.g., presence or absence) to quantitative data that rivals or even exceeds laboratory data depending on the sample type, processing and QA/QC (e.g., field XRF). The HEER Office currently requires laboratory-quality confirmation data for final decision making, especially for clearance of “clean” areas from further investigation. Field screening can be used to designate DUs (see TGM Section 3) for more detailed characterization and/or guide initial removal/remediation actions (refer to XRF case study in this Section). Field screening is best employed when the site contaminants of concern are known and a better understanding of the nature and extent of the contamination is needed for site characterization or for a removal or remediation response action. Particular attention should be focused on those portions of the site characterization and/or removal/remediation action that require documentation of contaminant levels below applicable EALs for final clearance (i.e., “Perimeter DUs”). Laboratory confirmation data will generally be required for these areas.
- Training: Many field screening methods employ specific kits or special field equipment, which requires properly trained personnel to conduct the testing. Standard operating procedures for the use of any field screening method(s) should be included as an Appendix to site-specific Sampling and Analysis Plans.
More information on topics discussed in this section can be found in Site Investigation Design and Implementation (see TGM Section 3) and in Data Quality Assurance and Quality Control (see TGM Section 10).
8.2 DATA QUALITY CONTROL AND DOCUMENTATION
Table 8-1 summarizes the data quality levels and objectives of “field screening” and “field analysis” data. Field analysis is used to discuss data quality control for quantitative field methods that can approach and even exceed laboratory quality data, though in many situations the cost of field analysis level data (with laboratory confirmation analyses) could exceed the cost of DU-MIS sampling with laboratory analysis.
| Table 8-1 Data Quality Levels for Field Screening and Field Analysis Methods | ||
| Data Quality Level | Purpose of Testing (Examples) | Example Methods |
| Screening – Qualitative or Semi-Quantitative |
|
Portable PID, VOC headspace analysis, in situ XRF, colorimetric analyses, and in situ sensor or probe equipment. |
| Field Analysis – Quantitative (with QC similar to a fixed laboratory analysis) |
|
Ex situ XRF, and many immunoassay, colorimetric, and turbidimetric kits, with laboratory confirmation data |
Field screening results can be presented in terms of presence or absence or in terms of relative concentration, for example above or below a pre-specified limit. The Photo Ionization Detector (PID), used to identify the presence and relative abundance of volatile organic chemicals (VOCs) in soil, is one of the most common field screening tools (see Subsections 8.4.2 and 8.4.5). Some PIDs can be used to measure both total VOCs in vapors emitted from soil as well as concentrations of a limited number of individual compounds (e.g., benzene). Another example of a field screening tool is a colorimetric field test kit for petroleum that is pre-set to a target concentration of TPH (e.g., 500 mg/kg). In some cases, screening may identify the total concentration of a family of contaminants but not concentrations for targeted, individual compounds needed for final decision making. This is true for some PAH field test kits. Due to potential matrix effects from moisture and large particles, the in situ use of a field XRF is considered “screening” level data, even though it reports specific concentrations of metals in soil.
Quality control procedures for the field screening methods typically consist of:
- Familiarity with the instrument operation and operations manual;
- Instrument calibration consistent with operations manual instructions; and
- Written documentation of calibration(s), any instrument maintenance, and data collected in the field.
Field analysis results are, in contrast, intended to match quantitative data that would be obtained from a laboratory. The objective of field analysis is to estimate the mean concentration of a targeted contaminant in a specific area and volume of soil (i.e. DU) and/or for an individual sample. Examples include immunoassay kits to test for PCBs or the use of an XRF to test processed, ex situ soil samples for metals. In the case of the immunoassay kits the mass of soil tested is similar to the correlative method used by a laboratory (e.g., ten grams). A single test may be adequate to represent the sample in terms of sampling theory, assuming that the sample is adequately processed and subsampled (see representative sampling information in TGM Section 4). Averaging multiple tests of a single sample will be required with use of a field XRF, however, since the mass tested during a single reading is relatively small (e.g. approximately one gram).
Data for discrete samples, while accurate for the specific mass of soil tested, are difficult to extrapolate to bulk sample submitted for testing or identify localized areas of contamination due to random, small-scale heterogeneity (see TGM Section 4 and XRF case study in Subsection 8.4.1). The collection of grid point samples of sufficient mass (e.g., 300+g) from multiple points within a small area (e.g. 1-2m²), rather than from single locations can help improve data representativenes and reduce this uncertainty. Note that such samples are sometimes referred to as “composites” by field workers, although in a strict sense of sampling theory (and in some regulatory applications) this term specifically infers the mixing of soil from otherwise separate DUs and its use as described above is discouraged (refer to Subsection 4.4.11). While potentially useful to help identify large-scale patterns of contamination and plan more detailed investigations or for initial identification and removal of areas of contaminated soil, HDOH considers this type of data to be adequate for screening purposes only. The designation of DUs and collection of MI sample data as described in Section 3 and Section 4 is recommended for final decision making.
Use of field analysis methods requires more attention and quality control in the field than screening methods, including:
- Calibration of instrument;
- Preparation of field standards using soil from the site or same soil type from near the site;
- Preparation of comparibility curves (e.g., XRF vs lab extraction data; updated as the project proceeds);
- Documentation of representative sampling/analysis methods
- Field replicates;
- Blank data;
- Documentation of data printouts or read-outs; and
- Field-laboratory data correlation.
Most field analysis method documentation includes a section that describes method precision and accuracy, as well as method limitations. These aspects of the method should be consistent with the goals established in the QAPP. The method of laboratory confirmatory analysis should also meet the project data quality objectives.
Traditional laboratory data are currently relied upon for final decision making purposes. Correlation analyses can be performed for field and laboratory confirmation data to utilize in data interpretation and decisions (if correlations are good). This might include, for example, correlation of the concentration of arsenic reported through use of ex situ field XRF analyses to USEPA Method 6010B (ICP) laboratory analyses (e.g., refer to discussion of Field XRF below). A minimum of ten to twenty samples should be submitted for laboratory analysis in order to carry out a comparability analysis of field screening data versus laboratory method data (see Subsection 8.2). Selected samples should span the range of metal concentrations estimated for the field samples and be processed and subsampled at the laboratory using MI procedures (see Subsection 4.2.6). Additional samples should be collected as needed to generate an acceptable correlation. Prediction lines that reflect 95% UCL and LCL confidence interval (or alternative values) should be added to the regression curve in order to assess the precision of an estimated value with respect to the target action level. For example, prediction lines might allow the ICP-equivalent concentration for a field XRF data point to be estimated within a range of +/- 25 mg/kg with a 95% confidence level. If the range of potential ICP concentrations predicted for an XRF data point spans both above and below the action level then a conclusion regarding the presence or absence of a potential environmental hazard (e.g., direct exposure risk) cannot be made for the specified degree of confidence. Certified laboratories are preferred or laboratories with equivalent documentation of QA/QC protocols. The proportion of field samples selected for laboratory confirmation will depend in part on project-specific data quality objectives.
Samples submitted to the laboratory for development of field versus laboratory data correlation analysis must be the same as used in the field or representative splits of the same material (see Subsection 4.2.2 on laboratory sub-sampling for guidance). Submittal of the entire sample that was analyzed in the field is preferable for cases where non-destructive field analysis methods are used (e.g., field XRF). Using a rigorous method to prepare any representative splits of samples is important in order to minimize subsampling error. This can greatly affect the correlation of field data to the laboratory data. Error due to variability in contaminant concentrations at the mass of soil analyzed may be unacceptable unless the same minimum (and representative) mass of soil is analyzed for both tests.
Because the minimum subsampling and analysis mass necessary to reduce fundamental subsampling/analysis error to a reasonable level is related to the maximum particle size in the sample, it is also important that the maximum particle sizes in the sample are known (to select the appropriate minimum subsampling/analysis mass) or that the samples are dried and sieved to a target maximum particle size before analysis in the field and lab, so a target subsampling/analysis mass can be selected. Typically, samples are air-dried and sieved to the < 2 mm particle size, before analysis in the field (e.g. ex situ XRF analysis) and laboratory, resulting in a target minimum subsampling/analysis mass of 10 grams. Due to the small mass typically measured by XRF analysis (~ 1 gram), averaging of multiple field XRF analyses (e.g. 10-20 analyses) across the same bulk sample will be necessary for comparisons to laboratory analyses that use a minimum of a 10 gram representative subsample mass for analysis (< 2 mm particle size samples). This same consideration for minimum subsampling/analysis masses for the maximum particle sizes in the sample may also be a significant issue for other types of field screening or field analysis methods, depending on the typical mass of sample the particular method/instrumentation utilizes, and the data quality objectives for the site investigation (see Section 4 for more information on representative subsampling issues).
Details of the sample processing, subsampling mass, and analytical mass used for the field and laboratory samples should be included in the reporting of field and laboratory sample correlation analyses, and uncertainties discussed. Replicate subsamples should also be used to assess the precision of data and subsampling error. Samples used in the correlation analyses should be selected from the lower, middle, and upper range of concentrations measured in the field analyses, as well as samples with contaminant concentrations at or near the site action levels, if any are found.
A comparison of laboratory and field analysis data can be carried out through either a simple correlation analysis or through a regression analysis, depending on the project objectives (e.g., refer to Yates et al, 2003). A correlation coefficient (r) is used to evaluate the strength of the relationship between field and laboratory data, within the limitations described above for error associated with subsampling of the parent sample. The correlation should be positive. The association between the two data sets strengthens as the coefficient approaches 1 (see Yates et al, 2003; Taylor, 1990). Coefficients less than 0.35 are generally considered to represent low or weak correlations. Coefficients values between 0.36 and 0.67 are considered to reflect modest or moderate correlations. Values between 0.68 and 1.0 are considered to be strong, with r coefficients at or above 0.90 considered to be very high.
The data plots should be reviewed to further interpret the nature of the correlation. A strong correlation but consistent variability in field versus laboratory data (e.g., field data consistently lower or higher) can be interpreted to indicate a true difference in the methods. A strong correlation with random variability (e.g., field data randomly higher or lower than laboratory data) could reflect heterogeneity in the soil and difficulty in obtaining representative splits of a sample for field versus laboratory analysis. The lack of a strong correlation between field and laboratory data does not necessarily indicate that the field screening data are “wrong.” This could instead simply reflect the presence of significant short-scale heterogeneity within the samples and error associated with processing and subsampling and/or variability and error in one or both test methods. Determining the exact source or sources of error will require additional studies and may or may not be cost and time beneficial for the subject site investigation.
Laboratory data are preferred for comparison to action levels or for use in risk assessments. If a strong, linear correlation exists between data sets (i.e., >0.68) then a regression analysis can be carried out to quantify this relationship and predict the laboratory-equivalent concentration of a chemical in soil based on field screening data (Yates et al 2003; see also Cutler 2009, 2011). This can significantly improve interpretation of field screening data and confidence in initial decision making. Combined with the use of DU and MIS investigation approaches and associated field and laboratory QA/QC this level of effort could be used to complete a site characterization investigation and/or site cleanup action based on field screening/analysis approaches alone, and avoid the delay and added cost associated laboratory data. The HEER Office may, however, recommend DU-MIS with laboratory analyses for final confirmation samples (after cleanup actions) in these cases. The use of field screening/analysis data in conjunction with or in lieu of laboratory confirmation data should be discussed with the HEER Office project manager on a site-specific basis and documented in the site investigation or removal/remediation report.
8.3 FIELD SCREENING METHODS AND DOCUMENTATION
Field screening methods with supporting documentation equivalent to that published by the USEPA for standard laboratory methods typically offer a higher level of data quality and reliability (e.g., SW-846, USEPA 2010). Documentation for the field screening methods describes the intended use, proper application, interferences, and overall performance in comparison to laboratory methods. The methods also list the required equipment, supplies, reagents and standards as well as information on safety, pollution prevention, and waste disposal. In addition, the methods typically provide a procedure that outlines required quality control, calibration, data analysis and result calculation. As described in Subsection 8.4 below and in the links to USEPA methods, documentation has been published by the USEPA for many but not all field screening methods. Field screening methods from a source other than the USEPA methods would ideally cover the same level of detail, but at a minimum, documentation for field screening methods should cover the following:
- Method Description: A summary of the method and instrumentation, including a list of target analytes and detection limits.
- Calibration: Typically the HEER Office prefers a daily 3-point initial calibration, although some methods may only require a monthly initial calibration. A mid-level continuing calibration standard which is analyzed every 20 samples is recommended; however, some methods may only require one such standard at the beginning and end of daily sample analysis. The method should include criteria as to what constitutes an acceptable initial and continuing calibration.
- Blank: The HEER Office prefers a baseline or a blank every 20 samples; however, some methods may only require a blank at the beginning of daily sample analysis. The method should include criteria as to what constitutes an acceptable blank.
- Corrective Action: The method should include corrective actions for failure to meet the criteria for initial calibration, continuing calibration and/or blanks.
Field screening method information should be presented in the form of a standard operating procedure (SOP) and included in the QAPP. Documentation of the field use of a field screening method should be recorded on established field data sheets which at a minimum include:
- Instrument: Type, maintenance, initial and continuing calibrations, blanks, and any failure to meet criteria along with the corrective action taken.
- Sample analysis: Date and time of analysis, sample identification, instrument reading, including data analysis and result calculation if applicable.
Example sources for field screening methods are provided in Table 8-2:
| Table 8-2 Sources for Field Screening Methods | |
| Source/Website | Comments |
| USEPA’s Field Analytic Technologies (USEPA, 2007), https://clu-in.org/characterization/technologies/ | Overview of multiple field analytic techniques, with links to the majority of documents included on the Clu-in web page |
| USEPA’s SW-846 Methods: https://www3.epa.gov/epawaste/hazard/testmethods/sw846/online/ | Use search function to locate specific field screening methods |
| USEPA’s Expedited Site Assessment Tools For Underground Storage Tank Sites (1997):https://www.epa.gov/sites/production/files/2014-03/documents/esa-ch6.pdf | 1997 overview of field methods for petroleum |
| USEPA ETV Program: https://www.epa.gov/remedytech/environmental-technology-verification-etv-program-site-characterization-and-monitoring | View information under subheading “Verified Technologies”; includes links to supporting documents. |
| ASTM International: https://www.astm.org/Standard/standards-and-publications.html | Use search function to look for specific method information. |
| Interstate Technology and Regulatory Council (ITRC): https://www.itrcweb.org/Guidance | See field measurement methods in the Triad Approach and Site Characterization and Monitoring topics |
| California Environmental Technology Certification (ETC) Program: https://dtsc.ca.gov/environmental-technology-certifications-program/ Due to state funding shortfalls, these certifications are no longer current; however, the website is still a potential resource on field screening methods. |
See ETC “Hazardous Constituents” List for substance-specific technologies |
8.4 FIELD SCREENING METHODS FOR SELECT CONTAMINANTS AND MEDIA
Information is provided below for examples of field screening methods (USEPA methods or state environmental program methods) that may be applicable for site investigations or removal/remediation work. For USEPA methods, check for the latest applicable method editions at the SW 846 methods website (USEPA, 2010). Other sources of field screening methods may be acceptable, as discussed in Subsection 8.3. Site investigation typically includes both horizontal and vertical delineation of contaminants and this should be considered when selecting applicable field sample collection and screening or analysis methods.
8.4.1 METALS
| Type of Contamination | Applicable Methods | ||
| Method Reference | Method Name | ||
| Metals | Soil and Sediment | USEPA 6200 | Field Portable X-Ray Fluorescence (XRF) Spectrometry for the Determination of Elemental Concentrations in Soil and Sediment |

Method 6200 (USEPA 2007g): Field Portable XRF Spectrometry for the Determination of Elemental Concentrations in Soil and Sediment
Method: XRF uses either sealed radioisotope sources or X-ray tubes (more current technology) to irradiate a soil sample. This incident radiation dislodges electrons from the innermost electron shells creating vacancies, which are filled by outer shell electrons. This rearrangement of electrons results in emission of X-rays (or X-ray fluorescence) that is characteristic of a specific atom and can be quantified. A single reading typically tests up to one-gram of sample, depending on the XRF being used. Source radiation and detector performance may vary by manufacturer and model. Field calibration standards, and calibration standards (or standard reference materials) should be employed for calibration and QA/QC measures. Those involved in sampling and testing should be trained in the use of the device and in radiation safety in accordance with manufacturer specifications and state radiation safety policy.
Detection limits vary from <10 to >200 milligrams per kilogram (mg/kg) depending on the instrument type and settings, the acquisition time used to collect a reading and the target element. The analyst should be trained in the use of the device, including XRF theory, instrument operation and calibration, detection limits, sample collection and processing, potential interferences and biases, QA/QC and data interpretation, and radiation safety (Kalnicky 2001; see also Geotek 2013,b). An overview of these issues is provided below but is not intended to serve as a replacement for formal training.
Due to advances in X-ray tube technology and difficulties in transporting radioactive materials, most field-portable XRF instruments currently use X-ray tubes. Some simple instruments have a few operating modes (i.e. alloy analysis, soil analysis, etc.) and built-in software, which provides real-time numeric concentrations for various elements. More advanced instruments allow flexibility in operational settings (tube voltage and current settings; filters for conditioning x-ray source, etc.) and collection of XRF spectra for later processing and analysis. Note that XRFs used in Hawai‘i must be registered in the State of Hawai‘i through completion of a Department of Health Radiation Facility License (https://health.hawaii.gov/irhb/radforms/). A copy of the XRFs license should be maintained by the user in the field, along with contact information for the specific entity that the XRF is registered under. Consultants who intend to bring XRF equipment in from out-of-state must ensure that the equipment is first registered and that a copy of the RFL license is included. If the specific type and model of equipment to be imported is unknown, then this can be noted on the form as “to be determined”.
XRF technology can provide comparable or higher quality data for total metal concentrations in soil in comparison to standard laboratory extraction/analysis methods, even when highly quantitative and accurate methods such as Inductively Coupled Plasma (ICP) are used by the laboratory to analyze the extract. This is primarily due to the incomplete extraction and corresponding under reporting of metals at the laboratory using common acid digestion methods (e.g., 3050B acid digest). For example, a comparison of XRF versus ICP data (Method 6010B) for soil samples collected as part of an internal HDOH project (HDOH, 2015) indicated consistently higher concentration of arsenic, copper, iron and zinc for the XRF data (average 30-60%). In some cases, however, the XRF reported a consistently lower concentration of a metal in comparison to the ICP data. In the cited example, reported concentrations of lead were similar but slightly lower (average -7%) for the XRF, possibly due to interference from moisture in the soil. The precision of laboratory data can be improved by using a more vigorous extraction method such as Method 3051A (microwave digestion). Both XRF and ICP methods have spectral overlap limitations that must be evaluated by an experienced operator of the analytical equipment.
The common laboratory USEPA 3050B acid digest method, though not as complete a digestion as stronger acid digest methods, could be considered by some to more accurately reflect “available metal” exposures for site receptors. Higher total metal analysis results for XRF and strong acid digestion methods in the laboratory can be due, in part, to metal concentrations that are bound tightly in the matrix of the soil particles and not readily bioavailable, even if the soil was ingested by a site receptor. Therefore, the choice of laboratory sample digestion methods for comparison to in situ or ex situ XRF metals data will affect the resulting correlation analysis and data interpretation, and should be considered/addressed in the site investigation DQOs and discussed with the laboratory. As in any investigation, systematic planning and clear site investigation objectives are an important part of field XRF screening investigations (refer to Sections 3 and 4 for further information). USEPA Method 6200 provides information relevant to data quality objectives specific for use of an XRF (USEPA 2007g).
Target Analytes: The number of metals that can be tested depends on the type of XRF used. Target elements of potential environmental concern at sites in Hawai‘i are typically limited to lead, arsenic and mercury (refer to Section 9). Lead is commonly found in soil impacted by lead-based paint, lead shot, and fill material that includes incinerator ash. Arsenic contamination is associated with the use of arsenic-based herbicides and can be very localized (e.g., at former pesticide mixing sites or from historic weed control around electrical substations) or widespread (e.g., due to past use for weed control in sugarcane fields). Mercury can be encountered at former sugar cane seed dipping facilities.
The need to test for and report other metals as potential contaminants of concern should be examined as part of the Systematic Planning process (TGM Section 3) and included in the site investigation work plan. Anthropogenic-related soil contamination associated with metals other than lead, arsenic, and mercury above levels of potential concern is rare in Hawai‘i. Note that naturally occurring concentrations of heavy metals in the iron-rich, volcanic soils of Hawai‘i can be significantly higher than typically found on the mainland US (e.g., cadmium, chromium, titanium, vanadium and zinc; see HDOH, 2012). These background concentrations of metals are tightly bound to soil particles (e.g., iron hydroxides) and do not pose a risk to human health and the environment (see also HDOH, 2016). Binding of arsenic residues to soil also significantly reduces the bioavailability and potential health risk posed by this contaminant (e.g., see Cutler 2011) and Cutler et al. 2013). In the case of arsenic, bioaccessibility testing data rather than total concentration data are used for human health risk and removal/remediation decision making purposes (HDOH, 2011c).
Advantages of Field-based XRF:
- In situ or ex situ analysis can be performed.
- Samples can be analyzed in minutes.
- Samples can be analyzed for multiple elements (metals) simultaneously.
- Many XRF instruments are portable and can operate in the field on battery power.
- Samples are not destroyed and can be re-analyzed or further analyzed in the lab by other methods.
- Simple sample preparation means little to no waste generation (i.e., no acids, solvents, etc.).
Limitations of Field-based XRF:
- Detection limits may vary significantly depending on the type of XRF instrument, x-ray sources, detection resolution, sample matrix, moisture content, and the metal being measured. Detection limits, depending on conditions and operating parameters can be higher than laboratory-based analytical methods, so project specific reporting limits should be evaluated to determine if XRF is appropriate, and what XRF instrumentation and operating parameters would work best.
- Physical matrix effects result from variations in particle size, uniformity and sample surface (ideally flat and smooth). If it is determined that physical matrix effects are impacting XRF measurements, these limitations can be managed by proper ex situ sample preparation including sample drying, sieving and increasing the number of points tested within a sample in order to both reduce and better represent variability within the sample.
- Moisture content can affect sample results. Field studies have indicated that the XRF fluorescence signal and calculated element concentration can be reduced by as much as 1 percent for every 1 percent increase in soil moisture (Cutler 2009). This limitation can be initially evaluated by pre-testing soil samples from the site under both field-moist and dried conditions. Potential interference from moisture can also be managed by uniformly drying (ex situ) samples before analysis (air or oven drying at <40°C preferred). Alternatively, moisture content can be determined in samples after field XRF analysis in field-moist condition, and results corrected for moisture effect (analogous to a laboratory reporting concentration in dry weight basis after applying a moisture correction). To perform such moisture correction, a calibration curve between XRF response and moisture content for each element of interest is required.
- Different metals can produce XRF spectral responses that interfere with one other because of peak overlap or absorption/enhancement phenomena. Most of these interferences are corrected automatically by modern XRF instrument software or post-acquisition software; however, they are best handled by calibration using site-specific calibration standards (SSCS), as described below. Hawaiian soils derived from volcanic materials contain high concentrations of iron (up to 20% by weight), titanium (up to 5%) and other heavy metals. The presence of these metals can greatly reduce fluorescence intensity of other elements (e.g., refer to Geotek 2013). For example, high iron can interfere with the accurate analysis of cobalt. Reported chromium concentrations can be falsely elevated in the presence of iron. High titanium can interfere with analysis for barium. Obtaining accurate element concentrations in high-iron soils (or sediments) requires use of calibration standards with similar atomic density, or better yet the use of SSCSs.
- Quantification of arsenic with elevated lead can be problematic due to fluorescence peak interference (USEPA 2007), as the dominant lead peak (L-alpha) emits at the same energy as the dominant arsenic peak (K-alpha). Lead concentrations can be readily determined from an alternate (secondary) peak; however, the arsenic secondary peak, depending on the instrument, might be too weak for quantification at concentrations of interest (e.g., below the Hawai‘i background concentration of 24 mg/kg). Newer XRF instruments and their robust built-in analysis software do an excellent job of quantifying lead and arsenic based on analysis of multiple fluorescence peaks. If analyzing samples for arsenic in the presence of lead, a subset of samples should be analyzed by laboratory methods to confirm XRF accuracy.
- A specific license, including a radiation safety course, is required to operate some XRF instruments that have radioactive source materials (hence most portable units now utilize x-ray tubes). Follow instrument-specific instructions to ensure proper operation of equipment. Persons using the instrument, or working in the vicinity of the instrument, should be trained on x-ray safety as per instrument guidance.
HEER Office recommendations (see also Geotek 2013b):
Calibration Standards
- Calibration standards (standard reference materials [SRMs]) are materials with similar chemical and physical properties as site samples, but with known concentrations of the elements of interest, analyzed in conjunction with site samples (unknowns) to allow calculation of element concentrations in unknowns. SRMs are often provided with rental XRF instruments, or can be obtained through entities such as National Institute of Standards and Technology (NIST).
- Some XRF instruments have pre-defined calibrations for “typical” soil materials built into “canned” instrument software (“fundamental” calibration). Results using these calibrations should be verified with laboratory testing of the same sample, taking into account potential laboratory processing/extraction error.
- Site-specific calibration standards (SSCSs) are also recommended in order to address differences in commercial SRMs (Standard Reference Materials) and local soils. Samples of soils from the study site (or a site with similar soil type) are collected prior to field work. Elements of interest are determined by laboratory analysis using EPA analytical methods (e.g., for most metals, acid digestion by 3050B [often incomplete digestion] or 3051 [more complete digestion since it employs microwave heating] and ICP analysis of digest by 6010 [ICPES] or 6020 [ICPMS]). Either contaminated site samples (unspiked) or spiked (element of interest added at known concentration) samples can be used as SSCSs. Spikes can be prepared, for example, by drying and sieving a sample (typically a site sample screened by XRF to have low concentrations of the element of interest) to an appropriate grain size (<2mm for most site assessments), spiking the sample with the element(s) of interest to generate a desired screening concentration, and then analyzing the sample by laboratory methods for verification of the spiked concentration. High-quality SSCSs can be prepared at the laboratory and analyzed using a nearly complete acid extraction (e.g., USEPA Method 3051) and Inductively Coupled Plasma Mass Spectrometry analysis (ICP-MS). Two or more spiked samples that cover the potential range of contamination anticipated are typically prepared for use as SSCSs in the field (e.g., 50 mg/kg, 200 mg/kg and 1000 mg/kg for arsenic). These SSCSs can subsequently be used throughout a study to either: 1) confirm instrument performance in the field or 2) create a calibration curve to correct instrument readings to final reported concentrations.
- The XRF should be calibrated daily during field work per manufacturer specifications using commercially available Standard Reference Materials (SRMs) such as those supplied by National Institute of Standards and Testing (NIST) or SSCSs. This typically includes soil standards with known metal concentrations (e.g., NIST 2010) but can also include manufacturer-provided stainless steel coins and other materials. Include a known, low-concentration sample to check the instrument reporting level for targeted metals (e.g., a lab-supplied sample or preferably a SSCS). Following the calibration, a blank should be analyzed to confirm negative control (e.g., NIST silicon blank). These materials are typically provided along with an XRF instrument if rented from a commercial vendor. The NIST metal standard or preferably a SSCS should then be used to confirm positive control.
- More detailed, calibration curves to improve the accuracy of field data can be developed by comparison of XRF and laboratory analysis data for splits of multiple samples across a range of metal concentrations (e.g., see Cutler 2009). The resulting data can be used to develop algorithms for adjustment of XRF-generated concentrations to equivalent laboratory method concentrations. A minimum of 3-4 SSCSs are recommended for initial development of a calibration curve (more if a good linear calibration curve is not achieved over the measurement range of interest).
- Caution is advised for comparison of field XRF data against laboratory data. Sample preparation (e.g., drying, sieving and subsampling), sample heterogeneity (e.g., inadequate number of sample points analyzed in field), and laboratory acid digestion methodology (e.g., Method 3050B versus 3051A) are far more likely to be the cause of conflicting results than the analytical method used to test the sample (XRF versus ICP).
- Samples from the site (or a site with similar soil type and climate setting) should also be collected (prior to the primary investigation) to test for matrix suitability and potential interferences. For example, soil moisture is known to interfere with XRF readings. Prior to the investigation, samples can be collected during rainfall conditions similar to anticipated conditions during the actual investigation. Tests made on both moist and dried samples can provide information on potential moisture interferences in the field. This can be used to calibrate the XRF prior to field screening to account for moisture interference.
Ex situ XRF Screening
- Ex situ screening of soil is likely to provide more representative and useful data than in situ analysis. In situ testing is prone to error due to small-scale random, small-scale variability of metal concentrations in soil as well as matrix interferences (refer to Subsection 4.1.2).
- A Decision Unit (DU) should be designated for a specific area of interest (e.g., suspect Spill Area DU or targeted Exposure Area DU; refer to Subsection 3.4). A Multi Increment sample consisting of 30-75+ increments should then be collected from the targeted DU and tested in the field using a portable XRF (see Subsection 4.2 and Section 5).
- Alternatively, a grid of “discrete” sample points can be used to help identify large-scale patterns of contamination and designate DUs for more detailed characterization (refer to Subsection 4.3). The collection of traditional 100-200g discrete soil samples from a single location is not recommended due to the uncertainty imposed by potential random, small-scale variability of contaminant concentration around individual grid points (see Subsection 4.1.2). As an alternative, large-mass discrete samples can be prepared by the collection of a minimum of 300g of soil from multiple points (e.g., 5-10+) within a short distance (e.g., one meter) of a grid point (see Subsection 4.3). This will improve the representativeness of the sample data for the subject grid point.
- Samples should be manually mixed to reduce variability prior to XRF testing, but be aware of separation of fines from coarser material. Coarse material should be removed prior to testing (e.g., >2 mm; plant roots, stones, etc.). If samples are dry enough, field sieving to the <2 mm size fraction and mixing can reduce small-scale variability and improve measurement accuracy. Potential interference from moisture can be addressed by drying samples before analysis (air or oven drying at ≤40°C preferred). Alternatively, the moisture content of field samples can be determined after analysis and results corrected, if a calibration curve between XRF response and moisture content for each element of interest has been determined.
- Spread the sample (typically in a plastic bag) to a thickness of 1 to 2 centimeters in order to allow equal access to all portions of the soil sample for X-ray analyses. A minimum of 10 to 20 random locations within the sample (representing a total of at least 10 grams of soil, assuming 0.5 to 1.0 grams tested per reading) should be tested until a reasonably consistent mean can be established that is unlikely to significantly change with additional testing (target a relative standard deviation of ≤20% to determine adequate number of readings). Test locations on both sides of the bagged, spread-out sample.
- Compare the average of the readings for a sample to action levels for decision making (refer to Subsection 3.2.1 and Subsection 3.4). Note that small-scale variability within an MI sample (or between discrete samples) is not important in terms of risk. HDOH EALs are likewise not intended for comparison to individual, discrete sample data for more than qualitative, screening level purposes (refer to Subsection 4.3).
- The following additional preparation and analysis procedures can be used to improve the quality and reliability of ex situ XRF data: 1) Prepare SSCSs to improve XRF measurement accuracy, 2) Inspect raw XRF spectra to ensure data acquisition is executed properly and assess potential interference from overlapping peaks (e.g., arsenic and lead), and 3) Grind (mill) samples to reduce variability if needed and practical for field screening (e.g., check variance of multiple analyses on subsamples of a larger sample to determine whether grinding may be necessary to reduce variability).
In situ XRF Screening
- In situ XRF measurements can be used for collection of semi-quantitative data to: 1) Identify potential metals of concern in soils or sediments, 2) Approximate extent of contamination on the project site and aid in the designation of DUs, and/or 3) Aid in determining locations for collection of MI samples for ex situ measurements.
- In situ samples generally consist of exposed surface or subsurface soils (i.e. excavation sidewalls) and are analyzed by XRF in place. User’s should be aware that single, in situ tests are highly prone to error associated with random, small-scale heterogeneity (i.e., variability over a few inches; refer to Subsection 4.1). Soil contaminant concentrations at the scale of an individual XRF reading can vary dramatically over short distances. Slightly offset, replicate measurements should be taken at each location to improve accuracy for the targeted sample location. Be aware that concentrations can also vary significantly and randomly over distances of a few feet, and patterns suggested by individual test points are also prone to error.
- Readings should be biased to areas of finer-grained soil (i.e., <2 mm particle size), and avoid taking readings on rocks or other large debris. Use a trowel or similar device to lift grass and organic material away from the targeted soil. A small piece of polyethylene film or similar material should be placed against the soil in order to protect the XRF detector. Detector window films (typically mylar) can easily rupture. The detector should not be used in any capacity with a ruptured window film. Care should be taken when replacing a window film to not allow soil or dust to enter the instrument.
- Ensure the XRF instrument can tolerate rain conditions, if encountered. Use a plastic bag or other rain protection for the instrument when needed in the field, or consider collecting ex situ samples and testing them at a fixed and protected location on- or off-site.
- Follow manufacturer specifications regarding acquisition time for the sample analysis (e.g., 30 second measurement typically recommended). Longer acquisition times can improve reporting levels, and may be desirable in certain situations.
- SSCSs are recommended if improved measurement accuracy is needed. If only relative element concentrations are required, default instrument calibrations or commercial standard reference materials (SRMs) can be used for calibration.
Additional Information:
Sample Type
Screening of Multi Increment samples can be useful for rapid assessment of contaminant levels in a targeted area (e.g., spill areas of suspected significant contamination or exposure areas for evaluation of direct-exposure risk, etc.) as well as guide designation of additional DUs for sample collection prior to demobilizing the field team. A minimum mass of 1 to 2 kilograms is recommended for MI samples. A minimum sample mass of several hundred grams is recommended for discrete samples. Sample collection for surface and subsurface soils is described in TGM Subsections 4.4 and 4.5.
Sample Drying
Air or oven dry (<40°C) the entire sample (typically) to reduce interferences from moisture and improve the accuracy of the XRF readings. Air drying in humid settings could leave 5-10% moisture in samples, with resulting XRF compound peaks depressed by a similar amount. Oven drying at 40°C produces similar moisture content as air drying in a low-humidity room (although more quickly). In either case, constant sample weight over time will indicate that the sample has been sufficiently dried. After drying, samples should be crushed or mixed by hand, as necessary, to break up aggregates and then sieved to collect the particle size fraction of interest (e.g., <2 mm particle size for most site investigation work).
Field Subsampling
If necessary, a subsample of the processed or unprocessed sample can be collected for ex situ XRF analysis. This will require additional sample handling and can increase the error in the estimated mean concentrations of metals in the original sample. However, subsampling before analysis may be desirable in some circumstances (e.g. to reduce drying time for very moist soils or minimize interference from large rocks or other debris in gravelly samples).
Physical mixing, drying, sieving and, if warranted, grinding a sample can help reduce variability, but complete “homogenization” of a bulk sample is rarely possible before subsampling and is difficult if not impossible to verify without extensive testing of the sample. Separation of fines during attempts to homogenize samples can lead to further bias in the collection of subsamples for testing. A representative subsample is collected with the use of Multi Increment sampling methods: approximately 30 small increments are collected from systematic random locations across the bulk field sample which has been spread out to a thin layer (see Subsection 4.2.6). Subsampling error (precision) should be estimated from the variance of replicate subsample analyses in terms of relative standard deviation (refer to Section 4). When the original bulk sample has been subsampled for XRF testing, that subsample should be returned to the bulk sample prior to the collection of additional (replicate) subsamples for testing.
XRF Analysis
Concentrations of elements of interest in site soils are best determined using empirical calibration and SSCSs. Known concentrations of elements and their spectra are entered into instrument software, or post-acquisition spectra processing software, and regression equations (calibration equations) are utilized to calculate concentrations of elements in unknowns (site samples). If there are only one or two elements of interest, and site samples and SSCSs are all similar, calibration equations and determination of concentrations can be handled “manually” in a spreadsheet program. This approach has been shown to be effective for analyzing arsenic in Hawaiian soils (Cutler 2011).
Site samples and SSCSs should be analyzed for elements of interest by optimizing acquisition parameters (if the instrument allows) including tube voltage and current, target X-ray conditioning filters and acquisition time. Multiple readings should be carried out on each sample until the user is confident of the range of targeted metal concentration present at the scale of the mass tested, with the average of the readings used for comparison to action levels. The minimum of readings necessary to estimate a mean metal concentration for a sample is site-and sample-specific. Based on HEER office staff experience a minimum of ten points is recommended in order to assess a target RSD of <20%. Significantly more readings might be necessary for soils with a high variability in contaminant concentration. Increasing the number of readings decreases errors in estimate of concentration means for the sample as a whole. Raw XRF spectra should be inspected to ensure data acquisition was executed properly. Software that allows overlay of multiple spectra can help flawed spectra to be identified and discarded prior to analysis for elemental concentrations.
Samples can be analyzed in various sample containers, such as plastic cups with mylar film windows or in thin plastic bags. If using plastic bags, the material should be analyzed with the XRF separately to confirm they do not contain elements of interest. Samples placed in plastic bags should not be later analyzed for phthalates or chemicals that the plastic could contain at significant concentrations.
XRF Field Analyses vs Laboratory Data Correlations
Multi Increment samples and fixed-laboratory data, including proper processing and subsampling of bulk samples, are normally required for final decision making purposes unless otherwise approved by HDOH. The entire bulk sample should be submitted to the laboratory for MIS-type processing and analysis. If a subsample is removed for XRF field screening then this should be placed back with the bulk sample before submittal for laboratory analysis.
A minimum of ten samples should be submitted for laboratory analysis in order to establish a correlation of field versus laboratory data (see Subsection 8.2. Selected samples should span the range of metal concentrations estimated for the field samples. Samples submitted to the laboratory to be processed and subsampled using MI procedures described in Section 4. This is the minimum number of samples recommended to establish the correlation of field versus laboratory data. For samples dried and sieved to a maximum particle size of < 2 mm, a 10-gram minimum mass should be representatively subsampled from bulk field XRF samples for laboratory correlation analyses (representative subsampling is typically conducted in the laboratory, but could be conducted in the field with a proper SOP). Samples to be used for XRF vs fixed-laboratory data correlation curves should be selected from the full range of concentrations measured by the XRF, and from different depth intervals, if applicable. They should also include samples with analyte concentrations at or near the site action levels, if present. For some projects, initial samples selected for field and laboratory correlation analysis could be used as the SSCSs during any subsequent phases of the investigation.
The results of the laboratory analyses and XRF analyses should be compared using linear regression analysis. If the measured concentrations span more than one order of magnitude, the data should be log-transformed to standardize variance, which is proportional to the magnitude of measurements. The HEER Office considers a correlation coefficient (r) for the regression of 0.75 or greater to be considered good quality (r²>0.56). If the r is 0.9 or greater (r²>0.81) the data indicates the correlation between the XRF data and the laboratory data are very strong (also see Shefsky, 1997). Ex situ XRF analyses are more likely to produce a strong correlation to laboratory analysis results than are in situ XRF analyses.
Concentrations of metals reported using an XRF are often higher than reported by laboratory extraction-based methods due to incomplete digestion of the soil at the laboratory. Metals in un-weathered igneous mineral phases present in the soil, in particular silicate minerals, may not be completely dissolved by the commonly used EPA Method 3050B, and the resulting laboratory data will not be representative of the “total” concentration of metals present in the soil (as is represented by the XRF measurements). A more robust EPA digestion method (e.g. 3051 or 3052) can be considered to increase the likelihood that the laboratory and XRF data will be comparable. Also, laboratory matrix spikes run on Hawai‘i volcanic soils when using Method 3050 may fail QC criteria due to strong binding to or interference from iron or other compounds. This increases the difficulty of assessing error associated with this digestion method and comparing field XRF vs laboratory results.
However, the use of alternative (stronger) lab digestion methods (e.g., Methods 3051 or 3052) and interpretation of the resulting data should be discussed with HDOH on a site-specific basis. The use of more stronger soil digestion methods could complicate comparison of the data to background metals in soils, since data reviewed for background levels were in part based on Method 3050 (see HDOH, 2012). In addition, the more commonly used digestion method (Method 3050) is also informally assumed by many risk assessors to represent worst-case estimates of the fraction of potentially bioavailable metals in soil. Bioavailability can be reassessed on a site-by-site basis for lead and arsenic through use of bioaccessibility tests, as described in Section 9 and the HDOH EHE guidance document (HDOH, 2016).
XRF Calibration Curves
Calibration curves for field XRF measurements based on site-specific calibration standards (SSCSs) as well as data comparisons to laboratory analyses of the same samples can be used to increase confidence in decision making in the field (see Cutler 2009). A relatively small number of SSCSs can produce a high-quality calibration curve in soils with a relatively low variability in terms of soil characteristics and metal concentrations. More samples (e.g., up to 10 or more) may be required to generate an acceptable regression line in some situations (e.g., USEPA 2007). If more than one soil type is present at a site (e.g., with different proportions of iron), separate sets of SSCSs for each soil type are recommended.
Use of XRF Data for Final Decision Making
It is feasible that the combined use of grids of samples to assist in DUs designation, thorough field QA/QC, replicate sets of data for select DUs, and strong correlation of field data with fixed laboratory data could allow final decision making at a site based primarily on grids of field XRF sample data in the absence of MIS confirmation data (refer to Section introduction). This is especially true for cases where random variability of contaminant concentrations in soil is anticipated to be low based on past experience at similar sites or previous studies at the subject site. The case study provided below approaches this idea, but uncertainty in the interpretation of seemingly isolated “hot spots” and “cold spots” and the mean concentration of arsenic in soil along the perimeter of heavily contaminated areas necessitates the collection of MIS confirmation samples prior to or following initial remedial actions. Although still considered “screening level” data, the thorough manner in which the data were collected significantly expedited the site investigation and allowed for initial remedial action plans to be developed. Conclusions drawn from the data and initial remedial actions are to be confirmed through a followup, DU-MIS investigation.
Investigation Report
The following information should be in included in a site investigation report when XRF data are collected:
- Printout of raw, XRF data for the project that includes calibration data and corresponding sample ID numbers (include as an appendix);
- Summary of ex situ sample collection methods including DU designation, number of increments collected, tools used, sample container, approximate sample mass, etc., and/or summary of in situ testing method used;
- Summary of field processing, including removal of large material, moisture testing, drying and subsampling, as applicable;
- Preparation of site-specific calibration standards, if used;
- Summary of XRF instrument utilized, including maker, beam strength used, calibration method, and other pertinent information;
- Summary of XRF testing method, including targeted metals, XRF read time, number of points tested, problems encountered in the field, etc.,
- Summary of XRF data and laboratory data comparison and, if prepared, calibration curves based on site-specific samples;
- Discuss how the XRF data were used for decision making in the field and will be used for any subsequent investigation work, and other information pertinent to the project.
Case Study – Arsenic at a Pesticide Mixing Site (see also Cutler 2009):
Elevated arsenic was identified in MI samples collected from a nine-acre, former pesticide mixing area. The past history of the site suggested that contamination was likely to be localized in the northwestern area of the property. In order to confirm and expedite identification of this area, ninety discrete soil samples with a mass of a few hundred grams each were collected in a grid pattern across the property and analyzed for total arsenic with a field-portable XRF. A subset of these samples was analyzed offsite for both total arsenic (USEPA Methods 3050B/6020) and bioaccessible arsenic (environmental hazards from direct contact to arsenic-contaminated soil are managed in the State of Hawai‘i by evaluating bioaccessible arsenic). From this data a correlation was developed between the total arsenic (measured by XRF) and bioaccessible arsenic. This allowed initial estimate of the area and volume of contaminated soil above bioaccessible arsenic thresholds, and better cost estimates for evaluation of remedial alternatives. Confirmation sampling using the DU/MIS process will be guided by the discrete sampling and field XRF analysis results.

The XRF data indicate a core area of arsenic contamination in the location of a known, former pesticide mixing area on the property. The model-generated sharpness of boundaries between “clean soil” and “contaminated” soil is misleading, however, and gives a false sense of resolution (see Section 4). As the mean concentration of arsenic in the soil situated between the heavily contaminated area and the apparently clean area begins to approach the target action level, smaller-scale variability in concentrations within the soil will begin to be exhibited in the form of seemingly isolated “hot spots” and “cold spots” above and below the target action level. These spots are “real” in terms of the soil samples tested, but cannot be accurately extrapolated beyond the mass of the soil tested with any degree of certainty, based on the discrete sample data alone. The identification of a hot spot or cold spot at any given location within this transition zone could instead be purely random in nature. If additional samples were collected a short distance away, then concentrations could be reversed, with “hot spot” areas turning “cold” and vice versa. Removal of isolated, sample-size hot spots is unlikely to reduce the overall, mean concentration of arsenic within this zone. Perimeter DUs should instead be designated for these areas and MI samples used to estimate mean contaminant concentrations for comparison to action levels (refer to Section 3).
This type of heterogeneity-derived variability is commonplace in attempts to generate isoconcentration maps from large amounts of discrete data. Such patterns are readily apparent in maps of background metals in soil published by the US Geological Survey (USGS 2014). As stated in that document:
“Soil geochemistry can vary considerably over (the distance between sample points), and this variation will not be accurately portrayed by the (isoconcentration) interpolation procedure. This study… (was) designed to reveal regional-, national-, and continental-scale geochemical and mineralogical patterns extending over thousands or hundreds of thousands of square kilometers. The resulting data sets are not appropriate for the accurate estimation of the concentration of a given element or mineral at a site (location) where a sample was not collected.”
8.4.2 PETROLEUM
| Type of Contamination | Applicable Methods | ||
| Method Reference | Method Name | ||
| Petroleum | Soil | USEPA 4030 | Soil Screening for Petroleum Hydrocarbons by Immunoassay |
| Petroleum | Soil | USEPA 9074 | Turbidimetric Screening Method for Total Recoverable Petroleum Hydrocarbons in Soil |
| Petroleum | Soil | RemScan (REMScan, 2014) | Infrared Spectrophotometry |
| Petroleum | Soil | USEPA Innovative Technology Verification Reports (USEPA, 2008) | Various (see reference), examples include Chemetrics, Inc. “Remediaid TPH Starter Kit”, and Sitelab Corporation’s “UVF 3100 TPH Analytical Test Kit” |
| PAHs | Soil | USEPA 4035 | Soil Screening for PAHs by Immunoassay |
| Petroleum | Soil | Maine DEP-SOP:TS004 | PID Bag Headspace Test |
| Petroleum | Soil | Maine DEP-SOP:TS004 | Oleophilic Dye Test |
Current field screening methods are not capable of separately reporting TPH aromatic and aliphatic fractions. If needed, contact the laboratory for more detailed information on these types of analyses.
USEPA 4030 (USEPA, 1996l): Soil Screening for Petroleum Hydrocarbons by Immunoassay
Method: Test kits are commercially available for this method and the manufacturer’s directions should be followed. The soil sample is extracted, filtered, and the sample extract and an enzyme conjugate reagent are added to immobilized antibody. The enzyme conjugate competes with the TPH present in the sample for binding to the immobilized anti-TPH antibody. The test is interpreted by comparing the sample response to a reference response.
Target Analytes: Depending on the testing product selected, results can be used to determine relative levels of TPH (low, medium, high) or whether the sample exceeds a threshold concentration (5, 25, 100, or 500 mg/kg).
Advantages:
- Samples can be analyzed quickly and on-site.
- Method is not affected significantly by moisture content or pH.
Limitations:
- Small mass of soil tested;
- The sensitivity of any immunoassay kit depends on the binding of the target analyte to the antibodies used in the kit. These kits are most sensitive to small aromatic compounds (ethylbenzene, xylenes, and naphthalene), while petroleum fuels tend to be dominated by aliphatic compounds.
- Non-TPH compounds such as chlordane and toxaphene show cross reactivity and can cause false positives.
- The sensitivity of the test is influenced by the nature of the hydrocarbon contamination and any degradation processes at the site. Although the response of the test to different sites will vary, the results within a site should be consistent and could be interpreted with comparison to laboratory TPH data.
- Organic and clay-rich soils may limit the effectiveness of soil extraction and may require longer extraction times than other soil types.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Laboratory confirmation data required for final decision making.
USEPA 9074 (USEPA, 2007h): Turbidimetric Screening Method for Total Recoverable Petroleum Hydrocarbons in Soil
Method: A sample of soil is extracted with a solvent mixture (primarily methanol), filtered, and added to an aqueous emulsifier development solution. As a result the hydrocarbons precipitate out and become suspended in solution. The sample is measured in a turbidimeter using a beam of yellow light at 585 nanometers (nm). Commercially available turbidimetric kits include the PetroFLAG™ system (see also USEPA 2001f).
Target Analytes: Broad spectrum of petroleum hydrocarbons in the range of 10 to 2000 parts per million (ppm).
Advantages: Samples can be analyzed quickly on-site, and at low cost. Most test kits do not require power or use battery-operated components.
Limitations:
- Small mass of soil tested;
- Potential under reporting of TPH concentration due to evaporation of volatile petroleum hydrocarbon mixtures such as gasoline during test procedure.
- Organic-rich soils can cause a positive interference as naturally occurring compounds become suspended in solution and/or cause a negative interference due to a reduced effectiveness of the extraction.
- Extraction may be difficult with clayey soils.
- Higher moisture content will bias sample results lower because there is a resultant dilution of the extraction solvent.
- The response of individual PAHs varied greatly from compound to compound and therefore use of this method to quantify individual PAHs is not recommended although quantification of PAHs as part of a larger hydrocarbon fraction, such as diesel fuel is recommended.
- Temperature has an effect on the suspension and it is important to recalibrate if temperatures change more than 10 degrees.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Laboratory confirmation data required for final decision making purposes.
RemScan (REMScan, 2014): Infrared Spectrophotometry
Method: This hand-held instrument uses diffuse reflectance infrared spectrophotometry to measure non-volatile TPH compounds (C10 to C36) in soils (see Figure 8-4). It was developed in collaboration with the (Australian) Commonwealth Scientific Industrial Research Organization (Forrester et al., 2012)). It is portable, hand-held, designed for regular field use, and makes direct surface soil measurements (either surface soil or along the surface of soil cores). This device is especially useful to screen for middle distillate fuels (e.g., diesel, kerosene, etc.) and heavier hydrocarbons in soil. Carbonates and natural organic matter can interfere with the infrared signature but this can be accounted for with site-specific calibrations. Moisture contents greater than 5% can also interfere with readings. Air-drying of samples is recommended for best accuracy.
Data Use: Screening.
Target Analytes: Total petroleum hydrocarbons, (C10 to C36) in soils.
Advantages:
- Portable, samples can be analyzed quickly on-site.
- Easy to calibrate in the field
- Minimal sample preparation (primarily just air-drying the surface of the soil, if >5% moisture content.
Limitations:
- Small mass of soil tested. Affected by soil moisture content – air drying before measurement is recommended.
- High cost for purchase (rental may be a more affordable option for limited use)
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Laboratory confirmation data required for final decision making.

Figure 8-4 RemScan handheld infrared spectrophotometry for testing of TPH in soil; produced by Ziltek Pty Ltd of Australia. |
USEPA Innovative Technology Verification Reports (USEPA, 2008): Note: Various field testing methodologies are evaluated and detailed in “Verification Reports”. For example, two field measurement technologies evaluated and judged reliable for TPH in soil include: 1) the Remediaid TPH Starter Kit (USEPA, 2001g) and 2) the UVF 3100 TPH Analytical Test Kit (USEPA, 2001g).
Remediaid TPH Starter Kit (USEPA, 2001g):
Method: Measurement based on a combination of the modified Friedel-Crafts alkylation reaction and colorimetry. Dichloromethane is used as a reactant and the solvent to extract petroleum hydrocarbons. Anhydrous aluminum chloride is used as another reactant. At least five grams of soil can be utilized per analysis.
Data Use: Screening.
Target Analytes: Aromatic petroleum hydrocarbons in soil. Includes analysis for a number of petroleum products/fractions, including gasoline, diesel, crude, lube oil, BTEX, and PAHs.
Advantages:
- Can measure a wide range of petroleum products
- Is not sensitive to common chlorinated solvents
- Portable and battery powered colorimeter
- Can be operated by one person with basic wet chemistry skills
Limitations:
- Provides little response for MTBE or Stoddard solvent
- Minor sensitivity to soil moisture
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Laboratory confirmation data required for final decision making.

UVF 3100 TPH Analytical Test Kit (USEPA, 2001h):
Method: Soil measurement utilizes an ultraviolet fluorescence spectrophotometer. The fluorometer uses a mercury vapor lamp as its light source. Petroleum compounds in soil sample are mixed and extracted with methanol, and the extract is transferred to a quartz cuvette that is placed in the fluorometer. 10 gram soil samples can be analyzed.
Data Use: Screening.
Target Analytes: The test kit can measure gasoline range organics (GRO) and extended diesel range organics (EDRO) separately, by selecting the appropriate emission filter that corresponds to the wavelength those fractions will fluoresce.
Advantages:
- Method detection limits below 10 mg/kg.
- Can be operated by one person with basic wet chemistry skills
- Non-petroleum compounds (e.g. chlorinated solvents, humic acid) do not interfere
Limitations:
- Portable, but needs power source (can use 110-volt or 12-volt outlet from automobile)
- Provides little response for MTBE or Stoddard solvent
- Minor sensitivity to soil moisture
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Laboratory confirmation data required for final decision making.
USEPA 4035 (USEPA, 1996m): Soil Screening for PAHs by Immunoassay
Method: A sample of soil is extracted and filtered using a commercially available test kit. The sample extract and an enzyme conjugate reagent are added to immobilized antibody. The enzyme conjugate competes with the PAHs present in the sample for binding to the immobilized anti-PAH antibody. The test is interpreted by comparing the sample response to a reference response.
Data Use: Screening.
Target Analytes: The test is most sensitive to three (phenanthrene, anthracene, fluorene) and four (benzo[a]anthracene, chrysene, fluoranthene, pyrene) ring PAH compounds and also recognizes most of the five and six ring PAHs listed in USEPA Method 8310. This test detects PAHs present at concentrations above 1 mg/kg.
Advantages: Samples can be analyzed quickly, on-site, and at relatively low cost.
Limitations:
- Small mass of soil tested;
- The sensitivity of the test is influenced by the nature of the hydrocarbon contamination and any degradation processes at the site. Although the response of the test to different sites will vary, the results within a site should be consistent and could be interpreted with comparison to laboratory PAH data.
- Field extraction of PAHs may be less effective than the extraction methods used in a laboratory and excessive amounts of oil in soil samples will interfere with the analysis of PAHs.
- Alkyl-substituted PAHs, chlorinated aromatic hydrocarbons, and other aromatic hydrocarbons (dibenzofuran) have been demonstrated to be cross-reactive with the immobilized anti-PAH antibody. The presence of these compounds may contribute to false positives.
- Kits may be damaged if frozen or if exposed to prolonged heat.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this screening method for each project. Given the uncertainties with the PAH analyses, laboratory confirmation is recommended.
Maine DEP (MEDEP, 2012): PID Bag Headspace Test for Volatile TPH compounds
Method: A photoionization detector (PID) uses an ultraviolet (UV) light source to break down chemicals into positive and negative ions that can then be measure with a detector (see USEPA Field Analytic Technologies link in Table 8-2; refer also to RAE Systems 2010). A soil sample is placed in an approved container and the volatile constituents are allowed to come to equilibrium. A minimum 200 gram sample is recommended. The headspace is measured with an isobutylene calibrated PID, and a result expressed in parts per million by volume (also see Subsection 8.6.4). Double layer, metalized polyester/low-density polyethylene bags are recommended as sample containers (e.g., Associated Bag Company Item #183-52) in order to minimize vapor loss during the recommended fifteen- minute equilibration period.
The Maine guidance recommends the following approach:
- Label and open the bags;
- Unfold the bottom gusset to facilitate a uniform headspace volume;
- Place appropriate mass of soil in metalized bag (recommended minimum 200 g);
- Close bag leaving uniform headspace.
- Knead samples (in closed bag) if needed to break up clumps;
- Shake bags for 30 seconds;
- Let stand for 10 minutes (out of direct sunlight) in order for the headspace to equilibrate with the soil;
- Knead/shake bags for additional 30 seconds;
- Let stand for 2 minutes
- Note: the PID reading must be made within 30 minutes of sampling to minimize loss;
- Open bag carefully and insert probe of calibrated PID one third to half way into bag (approximately 4 inches);
- Seal bag as much as possible around probe;
- Allow instrument to read until concentrations start to fall;
- Record highest sustained reading.
Headspace concentration readings must be kept within the PIDs linear range or use the data as minimum concentrations if over the linear range. Concentrations over the linear calibration range could saturate the detector and the instrument may not be able to distinguish between higher levels of volatile compounds. Readings above the linear calibration response range used for the particular PID should be estimated as above the upper calibration concentration utilized (different brands of PIDs could have different linear response ranges, or the documented linear response range could be limited by the calibration standards available).
Either MI or samples collected from individual points can be screened. The HEER office recommends that MI samples be screened when feasible, in order to ensure adequate coverage of the targeted DU area and better represent the results of final confirmation soil samples. If samples from single points are used, then soil should be collected and combined from multiple locations within the immediate vicinity of the sample point in order to reduce the effects of random, small-scale heterogeneity. An adequate number of samples to cover the targeted area should be screened based on professional judgment in the field.
The Maine guidance provides specific PID screening levels than can be used for final, site closure decisions. For example, based on studies conducted by that agency, 200 grams of soil with 25 mg/kg TPHg placed in a 2.5 liter bag should generate a headspace reading of approximately 40 ppmv (based on MiniRAE PID readings, other PIDs may differ). This may prove useful for general screening purposes in the field. Assuming a near linear correlation at low TPHg concentrations in soil, a PID reading of 160 mg/kg would presumably reflect the HEER office Tier 1 EAL of 100 mg/kg for TPHg. A lower PID screening level of 100 ppmv is recommended due to Hawaii’s warmer climate (Maine study carried out under controlled temperatures of approximately 70ºF). A PID screening level of 10 ppmv is recommended for soils impacted with middle distillate fuels (e.g., diesel and JP-8) or when the fuel type is unknown. This corresponds to anticipated lower vapor emissions from soils containing 100 mg/kg TPHd in comparison to gasoline-contaminated soils, due to a lower proportion of light-end aromatics and aliphatic compounds in these fuels (assumed 10% of gasoline vapor emissions; see also HDOH, 2012). Consultants report that PID readings below 10ppmv are unreliable for screening purposes and can in particular be biased high due to moisture.
The Maine guidance recommends testing of very small discrete samples under some closure scenarios in order to obtain accurate readings for high PID readings. For example, the guidance recommends the use of a five-gram sample (approximately one teaspoon) and PID reading of 1,500 ppmv (using a MiniRAE PID) in order to assess potential direct-exposure risk to workers with respect to a target soil screening level of 5,000 mg/kg TPHg (the correlation of PID readings and soil concentrations are not strictly linear between low and high TPH concentrations in the Maine study.) Use of a small mass of soil in the 2.5L headspace bag is intended to ensure that the PID detector does not become saturated. At this time, the HEER office has not adopted PID screening levels for final decision making purposes, so concerns of PID saturation and the use of a small sample mass are not applicable and screening of larger and more representative samples continues to be preferred (e.g., MI sample collected from excavation floor or targeted core interval).
Data Use: Screening.
Target Analytes: Gasoline-related volatile aromatic compounds. PIDs are not good indicators of total TPH levels in soil vapors because they do not respond well to aliphatic volatile compounds, which dominate vapors from petroleum fuels (refer to Section 7 and Section 9).
Advantages:
- Samples can be analyzed quickly, on-site, and at low cost.
- Amenable to testing of large samples, including MI samples.
- PID reading cleanup levels presented in Maine DEP guidance can be used for general guidance but collection of soil confirmation samples and comparison to HDOH EALs is currently required for regulatory concurrence.
Limitations:
- Applicable only for soils contaminated with gasoline. Can also be used to screen for diesel, kerosene, JP-8 or other middle distillate fuels in soil but PID response is significantly lower than for an equivalent concentration of gasoline due to low, aromatic content of vapors.
- Requires use of a container that minimizes loss of volatile chemicals during the equilibration period.
- PID requires periodic calibration in field.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Laboratory confirmation data required for final decision making.
Maine DEP (MEDEP 2012): Oleophilic Dye Test
Method: Soil is added to a sample bottle with water and a rapidly dissolving red or blue oleophilic dye. Contents are shaken vigorously. Released petroleum attaches to a polystyrene bead or to the walls of the container. Bead will turn pink or blue, depending on the dye use, if the concentration of petroleum in the soil exceeds the method detection limit (e.g. 500 ppm). Kits tested by Maine DEP include Oil-In-Soil and Oil-Screen-Soil but similar kits may be available from other venders. Red dyes typically most visible. Indigo blue kits available if soil color interferes with interpretation of red dye test kits. Recommended for testing of diesel, kerosene, JP-8 or other middle distillate fuels in soil.
Data Use: Screening.
Target Analytes: Oleophilic, petroleum hydrocarbon compounds (see also petroleum discussion in Section 9).
Advantages:
- Samples can be analyzed quickly, on-site, and at low cost.
- Results can be assessed in terms of “saturated,” “positive,” “slightly positive” and “undetected.”
Limitations:
- Marketed kits only allow testing of small masses of soil (e.g., ten grams of soil placed in 50 ml bottle); may not be practical for screening of MI samples (30 g mass typically tested by laboratory).
- Not applicable for gasoline (only) contaminated sites, heavy crude oils (Bunker C), or bituminous materials like asphalt or waxes.
- Mineral oil and motor oils may be detectable, but detergents in some synthetic oils can interfere with color development in the kits (i.e., potential for false negatives).
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method.
8.4.3 PCBS
| Type of Contamination | Applicable Methods | |
| Method Reference | Method Name | |
| PCBs | USEPA 4020 | Screening for PCBs by Immunoassay |
| PCBs | USEPA 9078 | Screening Test Method for PCBs in Soil |
| PCBs | USEPA 9079 | Screening Test Method for PCBs in Transformer Oil |

Figure 8-6 (A) PCB test kit components and (B) RaPID Assay immunoassay PCB test kits (Method 4020) in use at Kure Atoll (photo courtesy of Element Environmental). |
USEPA 4020 (USEPA, 1996n): Screening for PCBs by Immunoassay
Method: Test kits are commercially available for this method. A sample of soil and an enzyme conjugate reagent are added to immobilized antibody. The enzyme conjugate “competes” with PCB present in the sample for binding to immobilized anti-PCB antibody. The test is interpreted by comparing the response produced by testing a sample to the response produced by testing a standard(s) simultaneously.
Data Use: Screening.
Target Analytes: PCBs in soils and non-aqueous waste liquids present at concentrations above 5, 10, or 50 mg/kg.
Advantages: Samples can be analyzed quickly and on-site at a lower cost than typically charged by fixed laboratories.
Limitations:
- Small mass of soil tested (typically 5-10g);
- Detection limits for PCBs may not be adequate in either of the field screening methods to meet HDOH action levels. While useful in identifying areas of high concentration, areas with low-level concentrations may be overlooked.
- Poor extraction and correlation to laboratory sample data noted by some consultants for soils with a high clay or organic carbon content;
- Depending on the test kit used, chemically similar compounds (e.g., petroleum hydrocarbons) may exhibit cross-reactivity and produce a false positive test result.
This test method cannot differentiate between different Aroclor mixtures or individual congeners.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Laboratory comparison data required for final decision making purposes.
USEPA 9078 (USEPA, 1996o): Screening Test Method for PCBs in Soil
Method: This electrochemical method can be used to determine the amount of polychlorinated biphenyls (PCBs) contamination in soils if PCBs are known to be the sole organic halogens at the site. Chlorine is removed from the PCB molecule using an organo-sodium reagent. The method is designed to provide quantitative field results over a range of 2 to 2,000 microgram per gram (μg/g, equivalent to mg/kg) PCBs. The test can be useful when screening soil for disposal based on the TSCA-based limit of 50 mg/kg PCBs for municipal landfills. Caution should be used, however, when using results for decision making purposes on discrete samples rather than MI samples.
Data Use: Screening.
Target Analytes: PCBs in soils and non-aqueous waste liquids present at concentrations above 5, 10 or 50 mg/kg.
Advantages: Samples can be analyzed quickly and on-site. Most test kits do not require power or use battery-operated components.
Limitations:
- Small mass of soil tested (typically 5-10g);
- Reacts to all sources of organic chlorine and the presence of chlorine-containing compounds other than PCBs will cause the analyzer to produce false positive results (e.g., organochlorine pesticides such as Technical Chlordane).
- Iodine and bromine containing compounds will also affect results if present in significant quantities.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Laboratory comparison data required for final decision making purposes.
USEPA 9079 (USEPA,1996p): Screening Test Method for PCBs in Transformer Oil
Method: Method 9079 uses a colorimetric test kit to screen for PCBs in transformer oil at preset levels of 20, 50, or 500 mg/kg based on total organic chlorine detection. Samples are extracted in the field using an organo-sodium reagent. The resulting chloride ions in the sample are measured using a colometric indicator. Test readings typically calibrated to Arochlor 1242 (a lower chlorine content Arochlor) to provide conservative results. The test can be useful when screening transformer oil for TSCA-based limits for disposal options.
Data Use: Screening.
Target Analytes: PCBs in transformer oil present at concentrations above 20 mg/kg.
Advantages:
- Samples can be analyzed quickly and on-site. Most test kits do not require power or use battery-operated components.
- Results considered conservative, so concentrations near (but under) the set points typically provide a positive result, and limit false negative results.
Limitations:
- Water in the sample above 2% will cause low readings and cause a noticeable reaction with the sodium reagent.
- Can only be used for transformer oil, not for PCB contamination in other types of oils.
- Reacts to all sources of organic chlorine and the presence of chlorine-containing compounds other than PCBs will cause the analyzer to produce false positive results (e.g., organochlorine pesticides such as Technical Chlordane).
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Laboratory comparison data required for final decision making purposes.
8.4.4 DIOXINS
| Type of Contamination | Applicable Methods | |
| Method Reference | Method Name | |
| Dioxins | USEPA 4025m | Screening for PCDDs/PCDFs by Immunoassay |
| Dioxins | USEPA 4435 | Method for TEQ Determinations for Dioxin-like Chemical Activity with the Calux Bioassay |
Dioxins is a term used to refer to polychlorinated dibenzodioxin and polychlorinated dibenzofuran (PCDD/PCDF) compounds.
USEPA 4025m (USEPA, 2014b): Screening for PCDDs/PCDFs by Immunoassay
Method: This method uses a commercially available enzyme immunoassay (EIA) kit based on a polyclonal antibody specific for PCDD/PCDFs. The kit response correlates with total sample TEQ because the antibody responds to the 17 targeted PCDD/F congeners in approximate correlation with their toxic equivalency factors (TEFs). The kit response to a sample is a single result representing the aggregate of the individual congener responses. Congener separation is not required to use this kit, nor is it possible to generate any congener specific data with the kit.
The sample extraction and one step cleanup specified in the original Method 4025 allowed for only semi-quantitative screening at roughly 500 pg/g. That method remains viable, but the performance of the immunoassay has been improved by substituting a more conventional extraction and a simple and rapid coupled column cleanup. Quantitative analysis is possible to low pg/g levels in soils using the modified method. Detailed information is available from the immunoassay kit manufacturer, which also makes the sample preparation columns. The quantitative performance of this method has been validated by the SITE Program in two separate phases (USEPA 2008f). The effectiveness of the rapid sample preparation (separate from the immunoassay) is indicated by the fact that it is used by numerous labs for cleanup of sample extracts prior to analysis by Method 8290/1613B. Because of these improvements the immunoassay method using the coupled column cleanup is referred to as Method 4025m by the kit manufacturer and most users, to distinguish it from the original Method 4025 (Cape Technologies 2009).
Although an extremely useful screening method, EPA 4025 or its modified version (and other dioxin methods) should not be mistaken for a readily used field method. Mobilization into the field would likely require infrastructure comparable to mobilizing for the field execution of laboratory methods such as 8260, 8015, 8270 etc. (excluding expensive analytical instrumentation).
Data Use: Quantitative data at preselected decision levels (requires comparability analysis with GC/MS data; see Subsection 8.2)
Target Analytes: PCDD/PCDFs in soil to 5pg/g (ppt)(pg/g; equivalent to ng/kg or parts per trillion).
Advantages:
- Rapid testing of samples and reduced cost in comparison to Methods 8280 (or 8290).
- Direct reporting of TEQ dioxin concentration avoids need to adjust results with respect to congener-specific TEFs;
- Qualitative data as TEQ, comparable to Method 8290 in the range of the kit standard curve;
- Effective removal of most interferences by use of the coupled column cleanup, including heavy oils, PAHs, and other problem organics;
- Reduced cost in comparison to GC/MS analyses allows for the inclusion of larger number of QC samples.
Limitations:
- Common organic solvents such as hexane, acetone, and toluene are required for sample preparation;
- Method requires fume hood but can be done in small field lab (note that no liquid acids such as fuming sulfuric are used in 4025m);
- Sample processing and cleanup procedures may limit field use of the kits, although use in a laboratory setting can reduce costs in comparison to GC/MS methods.
HEER Office recommendations: The HEER Office recommends that dioxin levels be confirmed on 10% of the samples using laboratory GC/MS analyses (but at least 3 samples, whichever is greater). The GC/MS analyses should be conducted on samples with the highest-reported, bioassay TEQ dioxins results.
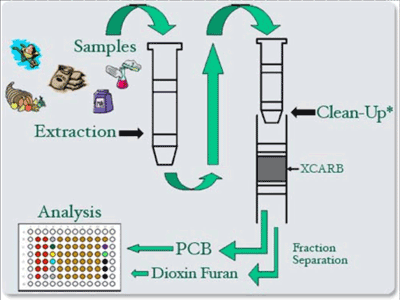
Figure 8-7.Schematic of sample processing steps for use of CALUX bioassay kit for Dioxins and PCBs (Xenobiotic Detection Systems, USEPA Method 4435). |
USEPA 4435 (USEPA, 2014c): Method for TEQ Determinations for Dioxin-like Chemical Activity with the CALUX Bioassay
In order to evaluate the accuracy and precision of this dioxin bioassay kit for soils in Hawai‘i, the HEER Office collected 25 multi-increment soil samples from a former sugar cane field in west O‘ahu and tested the samples for toxic equivalent (TEQ) dioxins using both High Resolution GC/MS and CALUX, USEPA Field Screening Method 4435 (HDOH, 2007e). The study showed that CALUX consistently over predicted TEQ dioxin concentrations in the soil in comparison to the GC/MS analysis. Dioxin TEQ concentrations estimated by CALUX ranged from 96% to 631% higher (average 297%) than Method 8290 data for splits of the MI samples. The difference was significantly higher for samples collected from an adjacent field, with CALUX data ranging from 416% to 1,346% higher (average 905%) than Method 8290 data for splits of the same samples (Tetra Tech 2012). Still other consultants have reported that CALUX produced slightly lower dioxin TEQ values in comparison to Method 8290 for soil with relatively low concentration of dioxins in soil (e.g., <50 ng/kg).
While the correlation of the CALUX test with the GC/MS data is somewhat low, the conservative nature of the CALUX test at concentrations above HDOH action levels supports its use as screening tool to estimate maximum levels of TEQ dioxins in soil. The consistency of the results suggests that the differences are more than random subsampling and laboratory error. This suggests that a pilot study on the use of CALUX methods and development of calibration curves prior to full-scale employment would be advisable.
Method: This method is a bio-analytical screening procedure for dioxin-like compounds in soils/sediments. This method is based on the ability of dioxin and related chemicals to activate the Ah receptor (AhR), a chemical-responsive DNA binding protein that is responsible for producing the toxic and biological effects of these chemicals. This method is a relatively rapid screening method capable of estimating the TEQs concentration for dioxin-like chemicals in a sample. A sample of soil is extracted in an organic solvent and fractionated through the sample processing procedure. An extract that contains the halogenated dioxins/furans is separated from an extract containing the halogenated biphenyls. These extracts are applied to monolayers of H1L6.1c3 cells and the amount of luciferase induction is measured after 20 to 24 hours. A standard dilution series of 2,3,7,8-TCDD is included on each plate of cells. Estimation of dioxin/2,3,7,8-TCDD-like TEQ activity present in the sample extract is performed by extrapolation to the 2,3,7,8-TCDD standard curve by least squares estimates with the 4 parameter Hill Equation. There are three modes by which the DIPS-CALUX bioassay is performed. These are the screening mode with historical recovery, screening mode surrogate recovery, and the semi-quantitative mode.
Data Use: Screening.
Target Analytes: PCDD/PCDFs in soil; a sample size of 2-10 g will typically give a detection limit of less than1 pg/g.
Advantages:
- Rapid testing of samples and reduced cost in comparison to Methods 8280 or 8290.
- Direct reporting of TEQ dioxin concentration avoids need to adjust results with respect to congener-specific TEFs.
Limitations:
- Small mass of soil tested (typically 5-10g).
- Field studies suggest overestimation of TEQ dioxins in comparison to GC/MS methods, especially at concentrations approaching or exceeding HDOH EALs;
- Samples expected to have extremely high levels of PAHs should be subjected to an additional cleanup step.
- Other contaminants in soil and sample extracts can be cytotoxic and impose a positive bias on the test (monitored as part of the method).
- Sample processing and cleanup procedures limit field use of the kits, although use in a laboratory setting can reduce costs in comparison to GC/MS methods.
HEER Office recommendations: The HEER Office recommends that dioxin levels be confirmed on 10% of the samples using laboratory GC/MS analyses (but at least 3 samples, whichever is greater). The GC/MS analyses should be conducted on samples with the highest-reported, bioassay TEQ dioxins results.
8.4.5 VOLATILES
| Type of Contamination | Applicable Methods | |
| Method Reference | Method Name | |
| Volatiles | USEPA 3815 | Screening Solid Samples for Volatile Organics |

USEPA 3815 (USEPA, 2007i): Screening Solid Samples for Volatile Organics
Method: A sample of soil is collected with minimal disturbance to minimize loss of volatile constituents using either a modified plastic syringe or commercially-available coring devices intended for this purpose. The sample is placed in a glass vial with organic-free reagent water, shaken, and a photoionization detector (PID) is used to measure the volatile organics found in the headspace over the sample/water mixture. This method may be especially suitable for screening of low levels of chlorinated solvents in soil. Refer also to field methods described for petroleum in soil in Subsection 8.4.2.
This method is strictly a screening procedure for estimating the total concentration of VOCs in soil and solid samples. This method can be used to estimate the relative concentration of VOCs in soil at a site prior to submittal to a laboratory and assist in the selection of low- or high-level analytical procedures at the laboratory (Hewitt and Lukash 1999). This can help ensure that adequate sensitivity can be achieved, while providing reasonable protection against overloading the analytical instrumentation used in quantitative purge-and-trap or headspace analyses. Use of this procedure may provide cost savings by minimizing the total number of aliquots of each sample that have to be collected for analysis by such quantitative laboratory-based techniques. Screening data can be compared to data for previously prepared, spiked soil standards from the site to estimate the concentration of VOC samples. The results of this screening procedure can also be used to guide other sample collection activities.
Data Use: Screening only.
Target Analytes: This method provides an estimate of the total concentration of volatile organics in soil. The method recommends using a standard at 200 ppb and using this standard to classify sample readings as “low” (below 200 ppb) or “high” (above 200 ppb).
Advantages: Samples can be analyzed in 5 minutes. Equipment is portable and battery operated. Sample analysis is cost effective.
Limitations:
- Small mass of soil tested (e.g., 5 grams);
- The type of volatile organics at the site must be known in advance and must be detectable by the PID. While PID responses to halogenated volatiles and aromatics may be fairly similar between compounds, the PID response to alkanes may be lower by an order of magnitude or more.
- As a screening method, this procedure is subject to a wide variety of potential interferences.
- False positives may occur if the PID is exposed to motor vehicle exhaust, solvents used for decontamination, or other sources of volatiles.
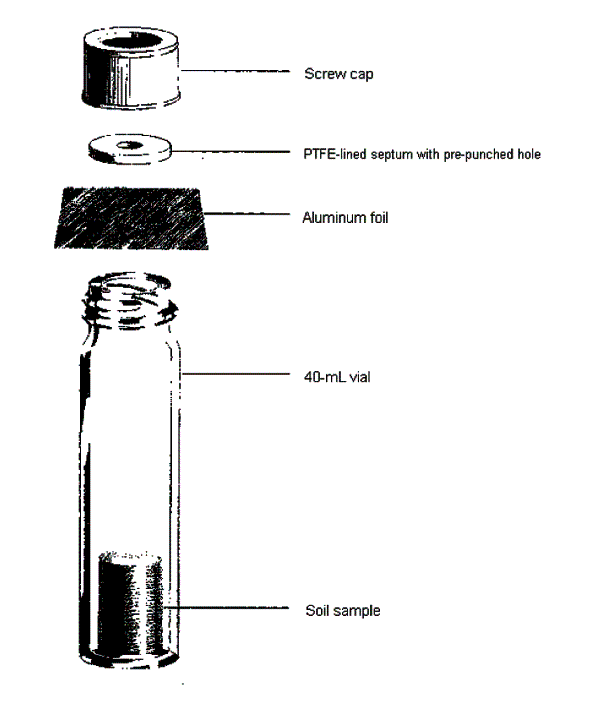
Figure 8-9 Container setup for headspace screening (Hewitt and Lukash, 1999) |
HEER Office recommendations: Headspace screening for chlorinated solvents in soil should be performed as specified in this method. Samples for VOC analysis should be collected immediately after the surface to be sampled has been exposed to the atmosphere (Hewitt and Lukash 1999). This includes samples both for screening and subsequent laboratory comparison analyses. The native structure of the material being sampled should experience minimal disaggregation during the collection and transfer process. The collection and transfer process should take less than 30 seconds and the sample weight is approximately (10 +2 g). The method specified sample container is shown above.
Immediately prior to analysis the vial should be hand shaken for 10-15 seconds for complete dispersion.
The most effective collection and handling protocols include either (a) the on-site, rapid transfer of discrete samples with a small coring tool to a hermetically sealable vessel that either already contains the appropriate dispersion/extractant solution or to which a solution can be added by puncturing the septum after a short period of storage (2 days at 4 degrees C) or (b) obtaining and temporarily storing (2 days at 4 degrees C) a sample in an EnCore sampler before transferring it to an appropriately prepared vessel.
Plastic bags or cores with plastic/Teflon on their end caps are not an acceptable substitution for the aforementioned sample collection methodology. Today it is recognized that past VOC sample collection and handling guidance often resulted in greater than 90 percent loss of VOCs from soil samples prior to laboratory analysis (Hewitt and Lukash, 1999).
8.4.6 PESTICIDES AND HERBICIDES
| Type of Contamination | Applicable Methods | ||
| Method Reference | Method Name | ||
| Pesticides | Soil | USEPA 4041 | Soil Screening for Chlordane by Immunoassay |
| Pesticides | Soil | USEPA 4042 | Soil Screening for DDT by Immunoassay |
| Pesticides | Soil | USEPA 4040 | Soil Screening for Toxaphene by Immunoassay |
| Pesticides | Soil, Water | USEPA 4015 | Screening for 2,4-D (in Soil or Water) by Immunoassay |
| Pesticides | Water | USEPA 4670 | Triazine Herbicides as Atrazine in Water by Quantitative Immunoassay |
| Pesticides | Soil, Water | USEPA 4010A | Screening for Pentachlorophenol (in Soil and Wastewater) by Immunoassay |
| Pesticides | Soil | USEPA 8540 | Pentachlorophenol in Soil by UV-Induced Colorimetry |

USEPA 4041 (USEPA, 1996q): Soil Screening for Chlordane by Immunoassay
Method: The method is performed using an extract of a soil sample. Filtered extracts may be stored cold, in the dark. An aliquot of the extract and an enzyme-chlordane conjugate reagent are added to immobilized chlordane antibody. The enzyme-chlordane conjugate “competes” with chlordane present in the sample for binding to chlordane antibody. The enzyme-chlordane conjugate bound to the chlordane antibody then catalyzes a colorless substrate to a colored product. The test is interpreted by comparing the color produced by a sample to the response produced by a reference reaction.
Data Use: Screening.
Target Analytes: Method 4041 is a procedure for screening soils to determine whether technical chlordane (chlordane isomers, endrin, endosulfan 1 and II, dieldrin and heptachlor) is present at concentrations above 20, 100 or 600 μg/kg. This analyte list is consistent with the HEER Office recommendation to evaluate total chlordane (see Subsection 9.1.3.3).
Advantages: Samples can be analyzed quickly and on-site. Most test kits require minimal training and are portable with battery-operated components. Sample analysis can be cost-effective.
Limitations: Compounds that are chemically similar may cause a positive test (false positive) for chlordane. In particular lower concentrations of aldrin and higher levels of toxaphene can cause a positive interpretation.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Multi Increment or equivalent sample data and laboratory comparison samples required for final decision making purposes. If compounds known to cause false positives are also present at the site, this method may not be appropriate.
USEPA 4042 (USEPA, 1996r): Soil Screening for DDT by Immunoassay
Method: The method is performed using an extract of a soil sample. Filtered extracts may be stored cold, in the dark. An aliquot of the extract and an enzyme-DDT conjugate reagent are added to immobilized DDT antibody. The enzyme-DDT conjugate “competes” with DDT present in the sample for binding to DDT antibody. The enzyme-DDT conjugate bound to the DDT antibody then catalyzes a colorless substrate to a colored product. The test is interpreted by comparing the color produced by a sample to the response produced by a reference reaction.
Data Use: Screening.
Target Analytes: Method 4042 is a procedure for screening soils to determine whether 1,1,1-trichloro-2,2-bis(4-chlorophenyl) ethane (DDT) (CAS Registry 50-29-3) and its breakdown products (DDD, DDE, and DDA) are present at concentrations above 0.2, 1.0 or 10 mg/kg. Method 4042 provides an estimate for the sum of concentrations of DDT and daughter compounds by comparison against standards.
Advantages: Samples can be analyzed quickly and on-site. Most test kits require minimal training and are portable with battery-operated components. Sample analysis can be cost-effective.
Limitations: Compounds that are chemically similar may cause a positive test (false positive) for DDT and its daughter products. In particular low concentrations of chloropropylate, chlorobenzilate, and dicofol can cause a positive interpretation.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Multi Increment or equivalent sample data and laboratory comparison data required for final decision making purposes. If compounds known to cause false positives are also present at the site, this method may not be appropriate.
USEPA 4040 (USEPA, 1996s): Soil Screening for Toxaphene by Immunoassay
Method: The method is performed using an extract of a soil sample. Filtered extracts may be stored cold, in the dark. An aliquot of the extract and an enzyme-toxaphene conjugate reagent are added to an immobilized toxaphene antibody. The enzyme-toxaphene conjugate “competes” with toxaphene present in the sample for binding to the immobilized toxaphene antibody. The enzyme-toxaphene conjugate bound to the toxaphene antibody then catalyzes a colorless substrate to a colored product. The test is interpreted by comparing the color produced by a sample to the response produced by a reference reaction.
Data Use: Screening.
Target Analytes: Method 4040 is a procedure for screening soils to determine whether toxaphene (CAS Registry 8001-35-2) is present at concentrations above 0.5 μg/g.
Advantages: Samples can be analyzed quickly and on-site. Most test kits require minimal training and are portable with battery-operated components. Sample analysis can be cost-effective.
Limitations: Compounds that are chemically similar may cause a positive test (false positive) for toxaphene. In particular low concentrations of endrin, endosulfan I and II, dieldrin, and heptachlor can cause a positive interpretation.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this screening method for each project. If compounds known to cause false positives are also present at the site, this method may not be appropriate. Laboratory comparison samples are required for final decision making.
USEPA 4015 (USEPA, 1996t): Screening for 2,4-D in Soil or Water by Immunoassay
Method: The method is performed using an extract of a soil sample, or directly on an aqueous sample. Filtered extracts may be stored cold, in the dark. An aliquot of the aqueous sample or extract and an enzyme-2,4-D conjugate reagent are added to immobilized 2,4-D antibody. The enzyme-2,4-D conjugate “competes” with 2,4-D present in the sample for binding to 2,4-D antibody. The enzyme-2,4-D conjugate bound to the 2,4-D antibody then catalyzes a colorless substrate to a colored product. The test is interpreted by comparing the color produced by a sample to the response produced by a reference reaction.
Data Use: Screening.
Target Analytes: Method 4015 is a procedure for screening soils and aqueous matrices to determine whether 2,4-dichlorophenoxyacetic acid (2,4-D) (CAS Registry 94-75-7) is likely to be present at concentrations above 0.1, 0.5, 1.0 or 5.0 mg/kg in soil, and 10 μg/L in water (ground water monitoring).
Advantages: Samples can be analyzed quickly and on-site. Most test kits require minimal training and are portable with battery-operated components. Sample analysis ican be cost-effective.
Limitations: Compounds that are chemically similar may cause a positive test (false positive) for 2,4-D. In general higher concentrations of chemically similar compounds are necessary to generate false positives and thus there are not significant interferences with this method.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Multi-increment or equivalent sample data and laboratory comparison data are required for final decision making purposes.
USEPA 4670 (USEPA, 2007j): Triazine Herbicides as Atrazine in Water by Quantitative Immunoassay
Method: This method uses a competitive immunoassay for the quantitative determination in water of triazine herbicides as atrazine. The method is performed using an aliquot of the water sample and an enzyme-atrazine conjugate reagent, which are added to an immobilized atrazine antibody. The enzyme-atrazine conjugate “competes” with triazine herbicides present in the sample for binding to the immobilized atrazine antibody. The enzyme-atrazine conjugate bound to the atrazine antibody then catalyzes a colorless substrate to a colored product. The test is interpreted by comparing the color produced by a sample to the response produced by a reference reaction.
Data Use: Screening.
Target Analytes: Method 4670 is a procedure for screening water with an optimal quantification limit of 0.03 μg/L for drinking water samples. The actual method sensitivity may be highly dependent on the kit used and sample matrix and the 0.03 μg/L lower detection limit may not always be achievable.
Advantages: Samples can be analyzed quickly and on-site. Most test kits require minimal training and are portable with battery-operated components. Sample analysis can be cost-effective.
Limitations: Compounds that are chemically similar may cause a positive test (false positive) for triazine herbicides. This cross-reactivity varies between different manufacturers and should be carefully evaluated for each project. Additionally, because this method quantifies triazine herbicides as atrazine, the response for the different compounds also varies between different manufacturers and also needs to be carefully evaluated for each project.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this screening method for each project. If compounds known to cause false positives are also present at the site, this method and/or specific kit for this method may not be appropriate. Laboratory comparison data is required for final decision making purposes.
USEPA 4010A (USEPA, 1996u): Screening for Pentachlorophenol (in Soil and Wastewater) by Immunoassay
Method: Method 4010A is a procedure for screening solids such as soil and sludge and aqueous media such as wastewater and leachate for pentachlorophenol (PCP) (CAS Registry No. 87-86-5). The method is performed using a water sample or an extract of a water sample. The sample/extract and an enzyme conjugate reagent are added to immobilized antibody. The enzyme conjugate “competes” with PCP present in the sample for binding to immobilized anti-PCP antibody. The test is interpreted by comparing the response produced by testing a sample to the response produced by testing standard(s) simultaneously.
Data Use: Screening.
Target Analytes: Method 4010A is recommended for screening samples to determine whether PCP is likely to be present at defined concentrations (i.e., kits are available which give positive results at 0.005 mg/L for aqueous samples, and at 0.5, 10 or 100 mg/kg in soil samples).
Advantages: Samples can be analyzed quickly and on-site. Most test kits require minimal training and are portable with battery-operated components. Sample analysis can be cost-effective.
Limitations:: Compounds that are chemically similar may cause a positive test (false positive) for PCP. In particular lower concentrations of 2,4,6-trichlorophenol and 2,3,5,6-tetrachlorophenol can cause a positive interpretation.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Multi Increment or equivalent sample data and laboratory comparison data are required for final decision making purposes. If compounds known to cause false positives are also present at the site, this method may not be appropriate.
USEPA 8540 (USEPA, 2007k): Pentachlorophenol in Soil by UV-induced Colorimetry
Method: The method was developed for wood treatment sites where PCP is the principal contaminant and other wood treatment chemicals, such as fuel oil and creosote are present. PCP is extracted from soil using methanol. An aliquot of the filtered methanol extract is added to acidified HPLC-grade water and the mixture is loaded onto a solid-phase extraction (SPE) column and eluted with hexane. The hexane eluate is mixed with basic water and is shaken. The aqueous solution is poured into a vial containing acidic water, octane, and cobalt chloride to facilitate separation. The mixture is shaken and allowed to separate. Approximately half of the octane is removed and added to a vial containing sodium sulfate. An aliquot of the octane is added to the vial containing the Quick Test® Reagent in an isopropyl alcohol solution. The mixture is placed in a plastic cuvette, mixed, and capped. The concentration of the PCP is determined using a dedicated colorimeter with the UV light exposure set at 260 nm.
Data Use: Screening.
Target Analytes: This method is recommended for analyzing soil samples to determine whether PCP is present at concentrations above 1.5 mg/kg. This method provides for the quantification of PCP relative to a three-point standard curve over a calibration range of 2 – 90 ppm.
Advantages: Samples can be analyzed quickly and on-site. Most test kits require minimal training and are portable with battery-operated components. Sample analysis can be cost-effective.
Limitations:
- Test reagents are highly sensitive to UV and have to be stored and used away from direct or indirect sunlight.
- The reagent can deteriorate slowly (based on post-exposure absorbance) but remains useful at 40 °C for up to 50 days. Deterioration at 60 °C occurred almost immediately, and post-exposure absorbance was significantly lower after 7 days. The reagent has been tested at 25 °C and remained stable for 4 months at that temperature.
- Compounds that are chemically similar may cause a positive test (false positive) for PCP; however, the clean-up steps in this method do eliminate most of this cross-reactivity and should not significantly impact this method.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this method. Multi Increment or equivalent sample data and laboratory comparison data are required for final decision making purposes.
8.4.7 EXPLOSIVES
| Type of Contamination | Applicable Methods | ||
| Method Reference | Method Name | ||
| Explosives | Soil | USEPA 4050 | TNT Explosives in Soil by Immunoassay |
| Explosives | Soil | USEPA 4051 | RDX in Soil by Immunoassay |
| Explosives | Soil | USEPA 8515 | Colorimetric Screening Method for Trinitrotoluene (TNT) in Soil |

Figure 8-11 Explosives immunoassay kit and explosives sample analysis using an immunoassay kit. |
USEPA 4050 (USEPA, 1996v): TNT Explosives in Soil by Immunoassay
Method: The method is performed using an extract of a soil sample. Samples and an enzyme-trinitrotoluene (TNT) conjugate reagent are added to an immobilized TNT antibody. The enzyme-TNT conjugate “competes” with TNT present in the sample for binding to the immobilized TNT antibody. The enzyme-TNT conjugate bound to the TNT antibody then catalyzes a colorless substrate to a colored product. The test is interpreted by comparing the color produced by a sample to the response produced by a reference reaction.
Data Use: Screening.
Target Analytes: Method 4050 is a procedure for screening soil samples to determine when TNT (CAS No. 118-96-7) is present at concentrations above 0.5 mg/kg.
Advantages: Samples can be analyzed quickly and on-site. Most test kits require minimal training and are portable with battery-operated components. Sample analysis can be cost-effective.
Limitations: Compounds that are chemically similar may cause a positive test (false positive) for TNT. In particular lower concentrations of tetryl, 1,3,5-trinitrobenzene, and 2-amino-4,6-dinitrotoluene can cause a positive interpretation.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this screening method for each project. If compounds known to cause false positives are also present at the site, this method may not be appropriate. Multi Increment or equivalent sample data and laboratory comparison samples are required for final decision making purposes.
USEPA 4051 (USEPA, 1996w): RDX in Soil by Immunoassay
Method: The method is performed using an extract of a soil sample. Samples and an enzyme conjugate reagent are added to immobilized hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) antibody. The enzyme-RDX conjugate “competes” with RDX present in the sample for binding to an immobilized RDX antibody. The enzyme-RDX conjugate bound to the antibody then catalyzes a colorless substrate to a colored product. The test is interpreted by comparing the color produced by a sample to the response produced by a reference reaction.
Data Use: Screening.
Target Analytes: Method 4051 is a procedure for screening soils to determine when RDX (CAS No. 121-82-4) is present at concentrations above 0.5 mg/kg.
Advantages: Samples can be analyzed quickly and on-site. Most test kits require minimal training and are portable with battery-operated components. Sample analysis can be cost-effective.
Limitations: Compounds that are chemically similar may cause a positive test (false positive) for RDX. In general higher concentrations of chemically similar compounds are necessary to generate false positives and thus there are not significant interferences with this method.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this screening method for each project. Multi Increment or equivalent sample data and laboratory comparison samples are required for final decision making purposes.
USEPA 8515 (USEPA, 1996x): Colorimetric Screening Method for TNT in Soil
Method: The method is performed using an extract of a soil sample. The sample is treated with color-change reagents and is read in a portable spectrophotometer. The colorimetric nature of the test is based on the visual detection of the reaction product that is formed when polynitroaromatic compounds react with acetone by ketone substitution in the presence of a base. This substitution product is measured at 540 nm using a spectrophotometer. The concentration of 2,4,6-trinitrotoluene (TNT) and closely related nitroaromatic compounds (2,4-dinitrotoluene, 2,6-dinitrotoluene, and 1,3,5-trinitrobenzene) in an unknown sample is determined by evaluating the intensity of the color that is developed.
Data Use: Screening.
Target Analytes: Method 8515 is a procedure for screening soil samples to determine when TNT (CAS No. 118-96-7) is present at concentrations above 1 ppm. The test also has similar reactivity to 2,4-dinitrotoluene, 2,6-dinitrotoluene, and 1,3,5-trinitrobenzene.
Advantages: Samples can be analyzed quickly and on-site. Most test kits require minimal training and are portable with battery-operated components. Sample analysis can be cost-effective.
Limitations: This test has similar reactivity for TNT as well as 2,4-dinitrotoluene, 2,6-dinitrotoluene, and 1,3,5-trinitrobenzene. Results from this test should take this fact into consideration.
HEER Office recommendations: The HEER Office recommends an initial site-specific evaluation of this screening method for each project. Multi Increment or equivalent sample data and laboratory comparison samples are required for final decision making purposes.
8.5 FIELD SCREENING WITH CONE PENETROMETER AND SENSORS/PROBES
There are a variety of sensors and probes that can be used to optimize sampling and analyses at contaminated sites. Geotechnical sensors can provide an indication of where historical fill materials could be present, and they can be used to refine information for processing geophysical data. In addition, geotechnical methods like cone penetrometer technology (CPT) can be used to obtain detailed geologic information, and high-resolution sensors/probes on on direct push platforms (DPT) can be used to delineate a water table and even to predict where vertical gradients could be present.
Other types of probes and sensors used in combination with CPT are designed specifically to target the identification of contamination in the subsurface, like the membrane interface probe (MIP) or fluorescence tools that look for hydrocarbons. These instruments are generally stacked together such that the maximum amount of information for a particular portion of a site is collected as efficiently as possible. These tools can be extremely valuable, but are also selective in terms of the type of data they can generate and the requirements for collecting data under controlled conditions.
Mobilization costs for sensors and probes with CPT can be significantly more than other sampling/analysis strategies, especially for small numbers of samples. However, for large numbers of samples appropriate sensors or probes in combination with CPT may be more cost effective and a valuable assistance to site delineation efforts.
8.5.1 DESCRIPTION OF CONE PENETROMETER TECHNOLOGIES
CPT techniques include samplers and analytical devices (typically a steel cone) that can be deployed into the subsurface using a direct push platform. Direct push platforms use hydraulic pressure to push a steel rod into the ground. This creates a small borehole by pushing soil out of the way as opposed to removing soil as in drilling. The same method is used to advance the sampling devices, geotechnical sensors, or analytical sensors associated with CPT. The sensors (attached to the tip of the cone) are connected to electrical cable running inside the hollow push rods, enabling the collection of data by a computer acquisition system at the surface. As such, “real-time” data can be collected using CPT techniques.
Two platforms are used for direct push technology (DPT). The first advances the tool string by the weight of the truck and supplemental steel weights and is known as a CPT. The second uses the weight of the truck aided by a rotary hammer and is known as the rotary hammer system. The CPT system is usually mounted on a 10-30 ton truck, while the rotary hammer system is mounted on pick-up trucks. The use of the name cone penetrometer system for the larger platform is misleading since CPT technologies can be deployed from both the CPT and rotary hammer platform.
The two platforms differ in scale of application and, to some extent, in the types of instruments and tools that they deploy. The devices developed for these platforms are: samplers for soil, soil gas and groundwater; geotechnical sensors for soil texture and hydraulic conductivity; and chemical sensor sampling techniques to detect petroleum, volatile organic compounds, metals and explosives. The sensors are connected to data acquisition devices mounted on the trucks.
CPT techniques are utilized for field screening only. Use CPT data to guide the placement of boreholes and selection of sampling locations. Collect samples for laboratory analysis to verify contaminant plume extent and contaminant concentrations.
8.5.2 CONE PENETROMETER DATA VERIFICATION
Always verify interpretation of CPT data by correlating it to site data. Correlate CPT data for soil type to lithological samples collected from the site to allow accurate interpretation of the CPT data (USEPA 2005).
Correlate conductivity (resistivity) data collected by CPT techniques to samples collected from the site. Conductivity varies with grain size but also with soil water content and ionic strength of the pore water or groundwater. Ionic strength of the groundwater can change due to contaminant content. Dense non-aqueous phase liquids (DNAPL) have a very low conductivity and can thus be detected by conductivity measurements (USEPA 2004c and 2005). Light non-aqueous phase liquids (LNAPLs) can also be detected; however, other methods are more efficient in locating LNAPL plumes (USEPA 2005). Because conductivity is influenced by soil type, water saturation, solute type, solute concentration and presence of non-aqueous phase liquids, the interpretation of the data is not straight-forward and requires calibration against site samples.
8.5.3 CPT ADVANTAGES AND LIMITATIONS
Cone penetrometer technology is deployed through either of two platforms of direct push technology (DPT).The advantages of direct push technologies (DPT) are:
- Deployment of in situ instruments allows rapid, real-time collection of data, which may be used to guide further drilling and sampling efforts, avoiding laboratory turn-around times and remobilization.
- Sampling and data collection may be faster than with traditional drill rigs.
- DPT does not generate large quantities of soil cuttings, thus reducing the amounts of investigation derived waste generated during the course of an investigation.
- Installation of micro-wells or pre-packed wells is substantially lower in cost than installation of permanent monitoring wells using traditional drill rigs.
- The rotary hammer system is deployed using smaller rigs, often pick-up truck mounted, and are therefore more mobile than traditional drill rigs. Smaller rigs can often access buildings or difficult to reach off-road locations. Also small rigs can be used in areas with overhead wires or other overhead hazards.
Cone Penetrometer Technology (CPT techniques) have the following limitations:
- CPT techniques can be used only in unconsolidated formations. Hard layers, partially cemented sediments, rocks and boulders limit the penetration. However, rotary hammer systems have rotary capability and can be used to penetrate concrete or other thin hard layers.
- CPT can often be advanced to depths greater than 100 feet, but cannot advance boreholes as deep as traditional augers can.
- Vertical changes in formation density limit the method. Hard layers encountered under soft layers may cause refusal, bending or breaking of the drilling rod.
- When the CPT system is mounted on trucks that weigh 20 to 30 tons, they are limited to locations with firm ground (the rotary hammer system is mounted on pick-up trucks with fewer limitations).
8.5.4 OTHER CPT INSTRUMENTS
Piezocones can be used to determine the hydraulic conductivity of subsurface soils and the depth to groundwater. These data can be used to identify potential contaminant pathways in the subsurface, or to aid in the selection of sampling locations (USEPA 2005).
Available chemical analytical sensors include: laser-induced fluorescence (LIF) probes and fuel fluorescence detectors (FFD), which can identify PAHs; and membrane interface probes (MIP) and SCAPS Hydrosparge™ systems for the detection of volatile organic compounds in soil and groundwater (USEPA 2004c and 2005).
USEPA evaluation of chemical sensors used with CPT has revealed that the sensor output does not correlate well with results obtained from laboratory methods (USEPA 1998; USEPA 1995). The HEER Office considers chemical data collected with CPT techniques as qualitative. In addition, the chemical sensors often detect classes of analytes rather than specific analytes.
Induced Fluorescence Tools
There are two basic delivery systems that can be used to detect hydrocarbons in the subsurface. One is a laser-induced fluorescence (LIF) set of tools and another is the fuel fluorescence detection (FFD) systems. Both provide a method for real-time, in situ, field screening of hydrocarbons in subsurface soil and groundwater. The technologies are intended to provide highly detailed, qualitative to semi-quantitative information about the distribution of subsurface petroleum contamination. LIF and FFD sensors are generally deployed as part of integrated mobile CPT systems that are operated by highly trained technicians familiar with the technology and its application. See ASTM, 2010 for a standard describing characterization of petroleum contaminated sites with LIF.
LIF and FFD systems can, with relative degrees of success depending on the tool configuration, detect gasoline, diesel fuel, jet fuels, fuel oil, motor oil, grease, and coal tar in the subsurface. The data can be used to guide an investigation or removal action or to delineate the boundaries of a subsurface product contamination plume prior to installing monitoring wells or taking soil samples.
There are currently four major induced-fluorescence systems available to private sector clients: the rapid optical screening tool (ROST) systems, the ultraviolet optical screening tool (UVOST), the tar-specific green optical screening tool (TarGOST), and FFD. Also, the Site Characterization and Analysis Penetrometer System (SCAPS) LIF system is one of several CPT-mounted sensors developed through a collaborative effort of the Army, Navy, and Air Force under the Tri-Services Program, but it is only available for federal facility projects. The ROST system was developed by Loral Corporation and Dakota Technologies, Inc., and is available commercially through Fugro, Inc. The UVOST and the TarGOST are available commercially from several vendors including Dakota Industries. All of these systems, while differing in some respects, are similar in their theories and methods of operation.
Advantages:
- The primary advantage of using LIF systems is their ability to provide real-time chemical and geological information while in the field. This data can reduce and focus the amount of physical sampling and laboratory analysis, as well as optimize monitoring well placement.
- Systems are capable of achieving 200 to 300 feet of pushes in a 10-hour work day.
- The vertical spatial resolution is near 2.0 cm, which allows small zones of contamination to be delineated that might be missed by conventional sampling protocols.
- No drill cuttings are produced with the system, saving the logistical requirement of handling drums of cuttings and eliminating disposal costs.
- The sample holes can be grouted as the push rod is pulled from the hole. Also, the push rod can be decontaminated remotely as it is retracted from the hole. All the decontamination fluids are containerized in the process.
Limitations:
- The operation of the fluorescence system takes considerable experience. It takes many days and numerous projects to become familiar with the operation of the technology. Operation of the technologies is provided as services by their respective vendors for this reason.
- Although these sensors provide information regarding the relative degree of contamination that closely matches reference method data, little direct, quantitative correlation has been found to individual or classes of petroleum compounds.
- The cost of the large, truck-mounted versions of these systems may be prohibitive for small-scale projects. However, recent advancements in the delivery systems and laser electronics are making fluorescence systems capable of tackling almost any size job more economically.
- Some maintenance of the CPT tools and the LIF sensors is required, and breakdowns can be expected on long-term projects. For example, downtime due to breakage of fiber optic cables and push rods, fogging of the sapphire window, and problems with the grout pump or decontamination unit may occur.
- These systems can only be used where direct push is feasible, such as in unconsolidated sediments. The sensors are limited to a depth of 50 meters because of attenuation in the optical fiber umbilical cord.
- Minerals such as calcite, naturally occurring organic matter, and man-made chemicals also can fluoresce, which may cause interference problems. Smearing and a memory effect on the sensor may occur when pushing through fine-grained sediments such as clays.
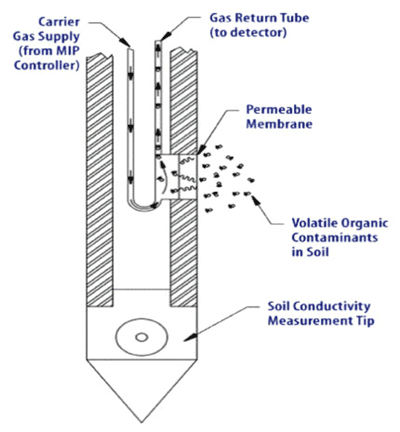
Figure 8-12 Membrane Interface Probe with Conductivity Probe Tip |
A MIP is a semi-quantitative field screening device that can detect VOCs in soil and sediment. It is used in conjunction with a direct-push platform (DPP), such as a CPT testing rig or a rig that uses a hydraulic or pneumatic hammer to drive the MIP to the depth of interest to collect samples of vaporized compounds. The probe captures the vapor sample, and a carrier gas transports the sample to the surface for analysis by a variety of field or laboratory analytical methods. Additional sensors may be added to the probe to facilitate soil logging and identify contaminant concentrations. The results produced by a MIP at any location are relative and subject to analytic verification.
MIP technology is capable of sampling VOCs and some SVOCs from subsurface soil in the vadose and saturated zones. It is typically used to characterize hydrocarbon or solvent contamination. Its ability to rapidly locate and identify contaminants reduces uncertainty in management decisions associated with costly cleanup projects, such as those commonly involving source zones of dense non-aqueous-phase liquid (DNAPL) and light non-aqueous-phase liquid (LNAPL). MIP technology uses heat to volatilize and mobilize contaminants for sampling. Heating the soil and/or groundwater adjacent to the MIP’s semi-permeable membrane volatilizes the VOCs, which then pass through the probe’s membrane and into a carrier gas for transportation to the ground surface.
The MIP consists of a small polymer (tetrafluoroethene) port, or membrane, that is permeable to gas but impermeable to liquid. The port is secured onto a steel block that also contains a resistive heater coil and a thermocouple, allowing the temperature of the membrane to be controlled and monitored. The heater coil heats the soil near the membrane to 80 to 125 ºC (160 to 232 ºF), which allows VOCs in the soil and groundwater to partition across the membrane in saturated or unsaturated soil. The subsurface temperature needs to be at or above the boiling point of the target compound(s). Nitrogen is the most commonly used carrier gas, but helium has been used in some applications. The carrier gas sweeps across the back of the membrane, entrains the VOC sample, and carries the VOC to the detection device located at the surface.
Typically, the MIP probe includes a tip that measures soil or water conductivity at a known distance below the membrane. The conductivity measurements can help correlate contamination to known soil stratigraphy. The probe conductivity measurements cannot identify the specific type of soil (based on grain size) distribution that is encountered unless the conductivity measurements can be compared to actual site soil core data. In the absence of on-site data, the MIP conductivity measurements identify changes in the soil’s electrical behavior that can be related to changes in stratigraphy or groundwater quality. Analytical devices commonly used with an MIP include gas chromatography (GC)-grade detectors (e.g., photo-ionization [PID], flame ionization [FID], electron capture [ECD], and dry electrolytic conductivity [DELCD] detectors) that establish the presence of VOC vapor, dissolved phase LNAPL, or DNAPL in soil. These detectors may be deployed singly or in line depending upon the site’s contamination. PIDs are best used for detecting aromatic compounds, such as BTEX (benzene, toluene, ethylbenzene, and xylene isomers). FIDs are used to detect petroleum hydrocarbons (straight and branched chain alkanes). ECDs and DELCDs are used to identify chlorinated hydrocarbons (e.g., PCE, TCE, dichloroethene, carbon tetrachloride).
Speciation of the contaminants can be accomplished either by collecting the off-gas on carbon or Tenax traps and subsequently desorbing the contaminants into a GC/mass spectrometer (MS), or by direct injection into an on-site ion-trap mass spectrometer (ITMS). Since the ITMS lacks a GC, its ability to resolve complex mixtures of contaminants is limited. See ASTM 2007b for a standard describing use of DPT for volatile contaminant logging with the MIP.
Advantages
- Real time data.
- Limited investigation derived waste.
Limitations
- MIPs provide screening-level data that need to be supplemented with analytical soil or groundwater data to fully support human health risk assessments or remediation decisions.
- Determining the depth at which the sample was taken when the sampler is in a near-continuous operating mode and the push rate is variable can be difficult. Compounds may be found in the subsurface for which the detectors were not calibrated.
- As with all direct push devices, MIP is only useful for deployment in unconsolidated matrices. Speciation with the ITMS can be problematic when the gas stream contains a complex mixture of chemicals. In many cases, the detection limit of MIP equipment for specific contaminants is above the detection limit required for human health risk assessment.
- ITMS-MIP overestimates contaminant concentrations for most vadose zone soils when compared with validation results, and it underestimates contaminant concentrations for clay-type vadose zone soils.
8.6 FIELD SCREENING EQUIPMENT TO SUPPORT HEALTH AND SAFETY PROGRAMS
The HEER Office does not regulate Health and Safety plans required for work on contaminated or hazardous waste sites (see Subsection 3.6.3). However, the HEER Office does check to confirm that Health and Safety plans are included as part of the overall site Sampling and Analytical Plan or site Work Plans. Because field screening equipment is commonly used on contaminated sites to support protection of workers, summary information on field equipment that may typically be utilized is included in this section.
All personnel using field survey equipment for health and safety related monitoring should have training on its operation, limitations, and maintenance. Maintenance and internal or electronic calibration should be performed in accordance with manufacturer recommendations by individuals trained and familiar with the devices before and after their use in accordance with manufacturers’ instructions. Repairs, maintenance, and calibration of these devices should be recorded in an equipment maintenance logbook. The equipment maintenance logbook for each instrument should be kept in that instrument’s storage case. For rented monitoring equipment, routine repairs consisting of maintenance and calibration should be conducted by the rental company vendor prior to the equipment being available for rent. The rental company vendor should provide a copy of the equipment maintenance and calibration response certification within the equipment storage case. The results of the vendor’s routine calibration and maintenance should also be recorded in the field logbook. For photoionization detector (PID) rentals, field personnel should ensure that the instrument contains the proper electron volt (eV) lamp.
Air monitoring equipment should be calibrated by field personnel before work begins and after each period of use, in accordance with manufacturers’ instructions and standard industrial hygiene practices to ensure the accuracy of the air monitoring data. Field personnel should ensure that they have the correct calibration gases for the intended air monitoring equipment to be used. Only basic maintenance (such as changing/charging batteries) will typically be performed by on-site personnel. Any additional maintenance or repairs should be performed by a trained service technician.
No single instrument can provide sufficient information as each has its own strengths and weaknesses. Any health and safety monitoring strategy should employ a combination of devices, be documented in an accident prevention plan and site-specific health and safety plan that meets applicable state and federal regulations and has been reviewed and approved by a certified industrial hygienist. Weather conditions such as extreme heat or cold, humidity (water vapor), exposure to rain or other spilled liquids, and electromagnetic radiation can all affect the instrumental readings for the equipment. Real-time monitoring should take into account the applicable use, operating ranges and limitations for the instrument being used, equipment warm-up time, equipment response time, and equipment correction factor (equipment sensitivity). Pay attention to inconsistent or non-responsive readings and record them in the field logbook. Air monitoring instruments discussed in the following subsections are typically used during soil and groundwater investigations. Additional air monitoring, radiation monitoring or other specialized equipment may be required.

Figure 8-13 Combustible Gas Indicator |
8.6.1 COMBUSTIBLE GAS INDICATOR (CGI)
This meter typically uses a platinum filament, which is heated by burning the combustible gas or vapor. The increase in heat is measured and reported as a percent of the lower explosive limit (%LEL). Generally if the gas is flammable, the Combustible Gas Indicator (CGI) reading is +50 percent when the meter is calibrated to hexane. The CGI has multiple sensitivities and thus can read a wide range of atmospheric levels on one meter. This meter operates only at normal oxygen levels and is also subject to electronic noise and is not very accurate at very low levels.
Advantages
- Measures the presence of combustible gases/vapors
- Range : 0 – 100 % of LEL (units are % of the LEL or ppm depending on the instrument brand used)
Limitations
- Catalytic sensor poisoning
- Relative response
- Does not indicate mists or dusts
- Must have normal oxygen content (generally at least 16 %) to provide valid readings

Figure 8-14 Oxygen Meter |
8.6.2 OXYGEN METER
This instrument uses an electrochemical sensor to measure the partial pressure of oxygen levels in the air and converts that reading to oxygen concentration. A deficient oxygen atmosphere (<19.5 percent) can indicate displacement by another gas or consumption. An oxygen atmosphere with a surplus (>23.5 percent) can indicate another source of oxygen. Either situation is potentially life-threatening and hazardous. It is important to understand that the meter only considers the effect of oxygen itself and not the presence of other materials. Any deflection on the oxygen meter should be treated as an abnormal situation. Neutralizing and masking gases can affect the accuracy of this instrumentation.
Advantages
- Measures oxygen levels in the air
Limitations
- Neutralizing and masking gases

Figure 8-15 Flame Ionization Detector |
8.6.3 FLAME IONIZATION DETECTOR (FID)
This meter has low detection levels for a broad number of organic compounds and has a self-adjusting span for known compounds. The FID ionizes compounds by burning them in a hydrogen flame. Operations on site may result in variable background levels of airborne compounds. Airborne compounds may be released from vehicles, blowing dust, material transfers, and so on. These sources can complicate monitoring of contaminant emissions during project tasks. Therefore, several upwind and pre-work measurements should be taken to assess contributions to airborne contamination by other potential sources. The instrument can also flame out at 10,000 ppm and above.
Advantages
- Low detection levels for many organic compounds
- Self-adjusting span for known organic compounds
- Ionizes compounds by burning them in hydrogen flame
- Can detect compounds with high ionization energy vapors (such as methane).
Limitations
- Can flame out at 10,000 PPM
- Multiple sensitivities
- Does not differentiate between compounds detected

Figure 8-16 Photoionization Detector (MiniRAE) |
8.6.4 PHOTOIONIZATION DETECTOR (PID)
This instrument detects atmospheric contaminants by ionizing them with UV radiation and producing a current that is proportional to the number of released ions. A PID with a 10.6 electron volt (eV) lamp should be sufficient for measuring VOCs and can be used for monitoring activities. A PID can determine compounds at very low concentrations with little electronic noise; the typical range is 0.5 to 2,000 ppm. The instrument is self-adjusting for known compounds but is blind to many common gases (methane, for example). Operations on site may result in variable background levels of airborne compounds. Airborne compounds may be released from vehicles, blowing dust, material transfers, and so on. These sources can complicate monitoring of contaminant emissions during project tasks. Therefore, several upwind and pre-work measurements should be taken to assess contributions to airborne contamination by other potential sources. This instrument is not accurate at high levels.
Advantages
- Can detect many analytes and organic compounds at very low concentrations
Limitations
- Not accurate when analytes and compounds are present in the air at high concentrations
- Does not differentiate between compounds detected
- Does not detect acid gases (HCl, HNO₃)
- Does not detect PCBs
- Humidity reduces accuracy
- Dusts / Mists reduce accuracy
- Extreme heat and cold may affect instrument response
- Does not detect methane and other vapors with high ionization energy
- Multiple sensitivities (multiple electron volt [eV] lamps may be needed)